Vibrational Spectroscopic Study of N-[4-[1-Hydroxy-2-[(1-Methyl Ethyl) Amino] Ethyl] Phenyl] Methane Sulfonamide
2016-06-15BalamuruganSampathkrishnanCharanya
N. Balamurugan, S. Sampathkrishnan, C. Charanya
1. Department of Physics, Sri Lakshmi Ammaal Engineering College, Chennai, Tamilnadu, India
2. Department of Physics, Sri Venkateswara College of Engineering, Sriperumbudur 602105, Tamil Nadu, India
3. Research Scholar, Department of Physics, Sri Venkateshwara College of Engineering, Sriperumbudur 602105, Tamilnadu, India
Vibrational Spectroscopic Study of N-[4-[1-Hydroxy-2-[(1-Methyl Ethyl) Amino] Ethyl] Phenyl] Methane Sulfonamide
N. Balamurugan1*, S. Sampathkrishnan2, C. Charanya3
1. Department of Physics, Sri Lakshmi Ammaal Engineering College, Chennai, Tamilnadu, India
2. Department of Physics, Sri Venkateswara College of Engineering, Sriperumbudur 602105, Tamil Nadu, India
3. Research Scholar, Department of Physics, Sri Venkateshwara College of Engineering, Sriperumbudur 602105, Tamilnadu, India
The vibrational spectral analysis was carried out by using FT-Raman and FT-IR spectroscopy in the range 4 000~400 and 4 000~400 cm-1respectively, for N-[4-[1-hydroxy-2-[(1-methyl ethyl) amino] ethyl] phenyl] methane sulfonamide (HPAEPMS) molecule. Theoretical calculations were performed by ab initio Density Functional Theory (DFT) method using 6-31G(d,p) basis set. The complete vibrational assignments of wavenumbers were made on the basis of potential energy distribution (PED). The results of the calculations were applied to simulated spectra of the title compound, which show excellent agreement.
FT-IR; FT-Raman; DFT; Vibrational analysis
Introduction
Generic name for N-[4-[1-hydroxy-2-[(1-methyl ethyl) amino] ethyl] phenyl] methane sulfonamide (HPAEPMS) is sotalol. Amides, sulfonamides and their derivatives has been the subject of investigation for many reasons. The Molecular formula for HPAEPMS is C12H20N2O3S. It belongs to C1 point group symmetry and soluble in water. The amides are important constituent of many biologically significant compounds. The chemistry of sulfonamides is of interest as they show distinct physical, chemical and biological properties. The sulfonamide derivatives are known for their numerous pharmacological activities, antibacterial, antitumor, insulin-release stimulation and antithyroid properties[1]. In addition, the unsubstituted aromatic/heterocyclic sulfonamides act as carbonic anhydrase inhibitors[2-3]whereas other types of derivatives show diuretic activity (high-ceiling diuretics or thiadiazinediuretics), hypoglycemic activity and anticancer properties[4]. Although sulfonamides are best known as bacteriostatic[5]and anti-malarial agents[6], there is now a range of drugs, possessing very different pharmacological activities, in which the sulfonamide group is present[7]. Several of these drugs suffer from bioavailability problems or adverse secondary effects[8-9]. Infrared, vibrational, NMR, and DFT investigation of N-[4-[2-[2-[4-(methanesulfonamido) phenoxy] ethyl-methyl-amino] ethyl] phenyl] methanesulfonamide[10].
In recent years, among the computational methods calculating the electronic structure of molecular systems, DFT methods has been favorite one due to its great accuracy in reproducing the experimental values of molecular geometry, vibrational frequencies, atomic charges, dipole moment, thermodynamical properties etc.[11-15]. DFT calculations are reported to provide excellent vibrational frequencies of organic compounds if the calculated frequencies are scaled to compensate for the approximate treatment of electron correlation, for basis set deficiencies and for the anharmooicity[16-19].
Literature survey reveals that to the best of our knowledge, no experimental and computational vibrational spectroscopic study free HPAEPMS is published in the literature yet. This inadequacy observed in the literature encouraged us to make this theoretical and experimental vibrational spectroscopic research based on the molecule to give a correct assignment of the fundamental bands experimental FT-IR and FT-Raman spectra on the basis of calculated Total Energy Distribution (TED). Therefore, the present study aims to give a complete description of the molecular geometry, molecular vibrations and electronic features of the present molecule. Besides, frontier molecular orbitals (FMOs) and thermodynamic properties were performed. Thermodynamic properties of the title compound at different temperatures were calculated.
1 Experimental details
The compound under investigation namely HPAEPMS is purchased from Sigma-Aldrich chemicals, USA with spectroscopic grade and it was used as such without further purification. The FT-IR spectrum of the compound has been recorded in Perkin-Elmer 180 spectrometer in the range 4 000~400 cm-1. The spectral resolution is ±2 cm-1. The FT-Raman spectrum was also recorded in the same instrument with FRA 106 Raman module equipped with Nd∶YAG laser source operating in the region 100~4 000 cm-1at 1.064 μm line widths with 200 mW powers. The spectra were recorded with scanning speed of 30 cm-1·min-1of spectral width 2 cm-1. The frequencies of all bands are accurate to ±1 cm-1.
2 Computational details
The entire calculations were performed at DFT (B3LYP) levels on a Pentium Ⅳ/1.6 GHz personal computer using Gaussian 03W[20]program package, invoking gradient geometry optimization[21]. This geometry was then optimized at DFT level, using basis set constraint 6-31G (d,p). The optimized structural parameters were using in the vibrational frequencies calculations resulting in IR and Raman frequencies together with Intensities and Raman depolarization ratios. The harmonic vibrational frequencies were calculated at the same level the optimized structures and obtained frequencies were scaled by 0.961[22]. It should be noted that Gaussian 03W package does not calculate the Raman intensity. The Raman activities were transformed into Raman intensities using RaInt program[23]by the expression:
WhereIiis the Raman intensity,Sis the Raman scattering activities,νiis the wave number of normal modes and denotes the wave number of the excitation laser[24].
3 Results and discussions
3.1 Molecular geometry
The molecular structure along with numbering of atoms of HPAEPMS is obtained from Gaussian 03 and GAUSSVIEW programs and is shown in Figure.1. The most optimized structural parameters (bond length and bond angle) calculated by DFT/B3LYP with 6-31G(d,p) are compared with X-ray and electron diffraction experimental data[25]in accordance with the atom numbering scheme given in Figure 1.
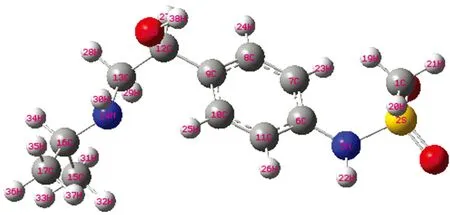
Fig.1 Numbering system adopted in the molecular structure of HPAEPMS
3.2 Vibrational assignments
The maximum number of potentially active observable fundamentals of a non-linear molecule, which contains N atoms, is equal to (3N-6) apart from three translational and three rotational degrees of freedom[26]. The present molecule HPAEPMS with 33 atoms and 93 normal modes of vibrations has C1 point group symmetry. The observed and simulated FT-IR and FT-Raman spectra of HPAEPMS are shown in Figures. 2 and 3, respectively. The observed and scaled theoretical frequencies using DFT/B3LYP with 6-31G(d,p) basis set with TED are listed in Table 1. Calculations were made for a free molecule in vacuum, while experiments were performed for solid phase. The vibrational analysis obtained for HPAEPMS with the unscaled B3LYP/6-31G(d,p) force field are generally somewhat greater than the experimental values due to neglect of anharmonicity in real system. These discrepancies can be corrected either by computing anharmonic corrections explicitly or by introducing a scaled field or directly
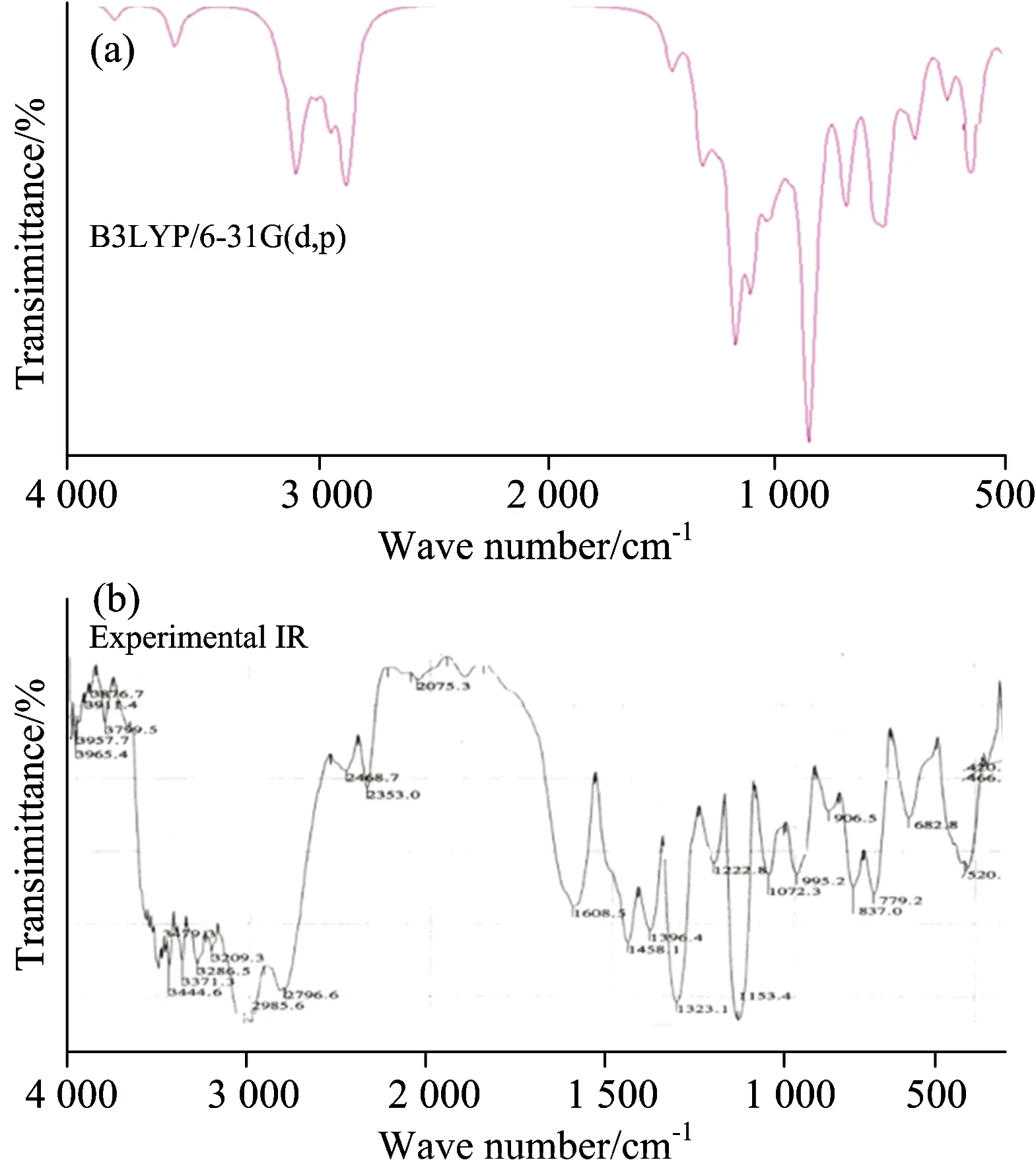
Fig.2 Comparative representation of FT-IR spectra for HPAEPMS
Table 1 The experimental FT-IR, FT-Raman and calculated frequencies using B3LYP/6-31G(p,d),) force field along with their relative intensities, probable assignments and Potential Energy Distribution (PED) of 1-N-[4-[1-hydroxy-2-[(1-methyl ethyl) amino] ethyl] phenyl] methane sulfonamide

ModesNoObservedfrequencies/cmFT⁃IRFT⁃RamanB3LYP/6⁃31G(d,p)UnscaledScaledIRRamanAssignments(PED)a13450(w)-3522347414 6493 61νNH(98)23444(w)-3588345037 2977 62νNH(98)33371(w)-351933833 6750 88νNH(97)43209(s)-323631111 21142 19νCH(97)5-3080(vw)323431091 0526 14νCH(94)63060(vw)3065(w)318830650 8753 01νCH(92)7--318330610 2970 11νCH(96)83055(w)3051(w)317730555 79117 22νCH(63)93050(w)3050(w)3174305121 2329 62νCH(97)10--3126300554 81113 84νasymCH3(100)113002(w)-3123300229 7650 89νasymCH3(96)12--3117299717 1126 76νasymCH2ofCH3(100)13--3107298731 5449 71νasymCH2ofCH3(100)14--3083296422 2841 54νasymCH2ofCH3(100)15--307729580 37121 68νasymCH2ofCH3(100)162928(m)2930(m)3047293011 69148 67νasymCH2ofCH3(100)172920(m)-3039292125 8461 45νasymCH2ofCH3(100)182850(w)-2987287269 7289 41νsymCH3(100)192800(w)2810(w)2932281993 08163 19Overtone+combination202796(w)-2909279771 1585 87Overtone+combination21--1666160143 50171 54νCC(55)+Ring2symd(16)22-1530(w)163115682 953 22νCC(42)+βCH(30)231490(m)-1552149290 432 19νCC(40)+βNH(16)24-1470(vw)152914702 912 29βCH(78)251460(s)-1525146626 807 35ρCH3(98)261458(w)1458(w)151514570 9721 12ρCH3(98)27--151014527 9011 57ρCH3(98)281440(s)1440150314451 1120 94δdefCH3(110)29--1490143219 806 4δdefCH3(100)301430(vw)1435(w)1488143023 644 86δdefCH3(100)31--147214154 5114 97βCH(64)+νCC(27)32-1410(w)146814121 6612 85ωCH3(75)331396(vs)-1430137518 872 22βCH(74)+νCN(26)34-1365(vs)14241369116 1035 99νasym(SO2)351360(m)-1421136627 609 28νs(SO2)36--1415136059 853 88δdefCH3(100)371345(vs)1340(w)1404134921 158 43νsymSO2(80)38--139013362 396 89Rind def (86)391332(vs)1337(vs)1384133027 328 63νsymSO2(66)+νCC(12)40--1359130711 490 95νasymSO2(66)+νCC(12)41--1358130518 357 71νasymSO2(66)+νCC(12)421300(vs)-13571304112 111 74βCH(12)+νsymSO2(66)43-1285(s)1337128520 192 74βCH(81)+νCN(19)44-1248(m)1303125363 2855 55νCC451222(m)1225(m)1279122965 9033 3νCC46--1247119928 1214 9βCH(47)+νCC(23)471195(w)-1246119823 1230 21βCH(47)+νCC(23)48-1185(vw)121911724 792 53γCH3(89)491160(vs)1211116427 3425 73νsymSO2(44)+νCC(21)501153(vs)1154(m)1208116119 0226 23βCH(45)+νCC(23)
To Table 1

511127(m)-1181113612 883 26νCC(28)+νsymSO2(15)+νCS(12)+νCN(11)52--116411199 282 82γCH3(66)53--1148110445 841 43γCH3(60)541092(m)1082(m)11341090262 683 75νasymSO2(50)+βCH(16)55--1117107451 415 7νasymSO2(79)561045(w)1015(m)1088104638 850 97Ringbreathing(630)57995(w)982(m)10379978 960 44γCH(92)58--100796831 245 34γCH(72)59-954(m)99095254 696 88γCH(79)60945(m)98594752 8311 74γCH(83)61--98094211 747 05γCH(80)62935(m)-9749362 240 37γCH(62)63--9649271 574 57γCH(83)64--9539170 423 51γCH(80)65906(m)904(m)94891212 585 71γCH(39)66-9358991 551 29ρSO2(49)+νC⁃SO2(16)67--88685198 539 24νCO(35)68837(w)-86583231 314 02γCH(33)69-817(m)85682319 066 97γCH(48)70800(m)804(m)83780588 4411 57γCH(45)71--83380110 552 75γCH(45)72779(m)780(m)82178919 1117 27βCCC(19)+τCCCH(14)73740(m)-76974028 146 18βCCC(19)+ρSO2(26)+νCS(15)74--7437149 732 5γCCC(87)+βCN(13)75698(m)-72169376 3210 53γNH(85)+βCN(10)76-665(w)69466811 546 75γNH(26)77-615(w)6466212 525 82τCCCC(23)+νCC(2)78575(vw)576(w)60057658 408 4τRing1asymd(38)79520(w)518(w)54152010 738 97βring(61)+νCC(21)80--50949054 745 03γCCC(44)+ρSO2(26)81475(w)479(w)49347475 219 03γCCC(44)+ρSO2(26)82--4864676 001 2τring(46)83466(w)-48246414 793 48τring(48)84-445(w)4564391 131 45γCCC(64)+ρSO2(27)85420(w)-4384213 540 92βCCC(47)+βCCO(16)86-403(w)4194032 520 18τCCCC(43)87--3883735 450 5βCCN(62)+rSO2(38)88-365(m)36635212 062 53βCCN(60)+rSO2(40)89--3363237 681 46βCCO(47)+βCCC(21)90--3213087 812 55βCOC(28)+βCCC(22)91--301290107 335 62βCOC(38)+βCCC(22)92-280(w)2972862 182 54τCCCC(45)93--2722622 281 11βCSN(16)+νCS(11)+rSO2(39)94--2572470 300 78βCSN(26)+νCS(10)+rSO2(40)95--2472372 852 32βCSN(25)+βCSO(17)96--2292201 540 05βCSN(35)+βCSO(27)97-206(w)2142061 471 14γ(ring⁃SA+ringSO2)(92)98--2011941 200 56νCS(36)+βCCC((14)+νCN(10)99-186(w)1931861 000 76γ(ring⁃SA+ringSO2)(80)100--1631560 640 47γ(ring⁃SA+ringSO2)(82)101-124(vw)1331282 460 57Ringbutterfly(84)102-103(w)1101061 090 84Ringbutterfly(88)103-78(m)92884 520 75τSO2(67)+τC⁃SA(17)104--50481 562 54τCH3(38)+τRing1symd(10)105--46441 920 72τCH3(74)+τRing1symd(15)
To Table 1

106--36351 321 95τSO2(85)107--27261 352 47τSA(93)108--14141 930 74τ(ring+SO2)(99)
aνsym: symmetric stretching;νasym: asymmetric stretching; symd: symmetric deformation; asymd: asymmetric deformation;β: in-plane-bending;γ: out-of-plane bending;ρ: scissoring;ω: wagging;r: rocking;τ: torsion; SA: sulfonamide; TED: Total energy distribution
scaling the wavenumbers by proper factor. A tentative assignment is often made on the basis of the unscaled frequencies by assuming the observed frequencies so that they are in the same order as the calculated ones. A better agreement between the computed and experimental frequencies can be obtained by using different scale factors for different regions of vibrations.

Fig.3 Comparative representation of FT-Raman spectra for HPAEPMS
3.3 N—H vibration
The high frequency region above 3 000 cm-1is the characteristic region for the ready identification of C—H, O—H and N—H stretching vibrations. In this region, the bands are not affected appreciably by the nature of substituents[26]. The investigated molecules have NH2group. Hence, in NH2group, one symmetric and one asymmetric N—H stretching vibrations are expected. It is stated that the N—H stretching vibrations occur in the region 3 300~3 500 cm-1[27]. Specifically, the asymmetric NH2stretching vibration appears from 3 420 to 3 500 cm-1and the symmetric NH2stretching is observed in the range 3 340~3 420 cm-1[27]. In this study, the FT-IR bands at 3 450, 3 444 and 3 371 cm-1have been assigned to NH2asymmetric stretching vibrations. The scaled vibrations calculated at 3 474, 3 450 and 3 383 cm-1by B3LYP/6-31G(d,p) method (mode nos. 1 and 3) which are listed in Table 4.2 correspond to asymmetric stretching mode of N—H units with the PED contribution of 98% and 97%, respectively. The N—H out-of-plane bending vibrations are observed at 698 cm-1in FTIR spectra. The bands observed at 665 cm-1in FT-Raman spectra have been assigned to N—H out-of-plane bending vibrations. The experimental values are found to be in good agreement with the theoretical values.
3.4 C—H vibrations
The aromatic structures show the presence of C—H stretching vibrations in the region 3 100~3 000 cm-1which is the characteristic region for the ready identification of C—H stretching vibrations. In this region, the bands are not affected appreciably by the nature of substituents[28-29]. The modes (4~9) are due to C—H stretching of hydrogen bonded carbon atoms of phenyl rings. These modes are pure C—H stretching vibrations with a PED contribution nearly 97%. In this study, the CH stretching modes of studied compounds were recorded in both FT-IR and FT-Raman spectra. The corresponding modes were observed at 3 209, 3 060, 3 055 and 3 050 cm-1in FT-IR and FT-Raman spectra were observed 3 080, 3 065, 3 051, and 3 050 cm-1. The scaled vibrations calculated at 3 111, 3 109, 3 065, 3 061, 3 055 and 3 051 cm-1by B3LYP/6-31G(d,p) method. The aromatic C—H in-plane bending modes of benzene and its derivatives are observed in the region 1 300~1 000 cm-1with a weak intensity in the vibrational spectra[29-30]. The C—H out-of-plane bending vibrations occur in the range 1 000~750 cm-1in the aromatic compounds[29-30]. The computations suggest that the expected other bands of C—H in-plane or out-of plane bending vibrations are masked by other strong vibrational modes. The same trend is observed in the title compound.
3.5 Methyl group vibrations
The molecule HPAEPMS under consideration possesses one CH3unit which lies in the terminal group of the molecule. For the assignments of CH3group frequencies, nine fundamentals can be associated to CH3group[31]. The C—H stretching in CH3occurs at lower frequencies than those of aromatic ring (3 100~3 000 cm-1). Moreover, the asymmetric stretch is usually at higher wavenumber than the symmetric stretch. Methyl group vibrations are generally referred to as electron-donating substituent in the aromatic ring systems, the antisymmetric C—H stretching mode of CH3is expected around 2 980 cm-1and CH3symmetric stretching is expected at 2 870 cm-1[32]. The first of these results from the antisymmetric stretching of CH3mode in which the two C—H bonds of the methyl group are expanding while the third one is contracting. The second arises from the symmetric stretching in which all the three C—H bonds expand and contract in phase. The CH3symmetric stretching mode is predicted by B3LYP/6-31G(d,p) method at 2 872 cm-1and it shows 100% TED contribution suggesting that it is a pure stretching mode. There is a peak observed at 2 850 cm-1for CH3 stretching vibration in FT-IR.
For methyl substituted benzene derivatives, the antisymmetric and symmetric deformation vibrations of methyl group normally appear in the region 1 465~1 440 cm-1and 1 390~1 370 cm-1, respectively[33-34]. The band at 1 460, 1 458 and 1 440 cm-1in FT-IR and 1 458 and 1 440 cm-1in FT-Raman are attributed to CH3scissoring vibrations. The wavenumber at 1 410 cm-1in FT-Raman and 1 406 cm-1in FT-IR is assigned to CH3wagging vibration.
3.6 SO2vibrations
The asymmetric stretching for the SO2, NH2, NO2, CH2and CH3, etc. has magnitude higher than the symmetric stretching[35-36]. The symmetric and asymmetric SO2stretching vibrations occur in the region 1 125~1 150 cm-1and 1 295~1 360 cm-1, respectively[37]. The intense signals appearing at 1 360, 1 345, 1 332 and 1 300 cm-1(IR) and 1 365, 1 340 and 1 337 cm-1. In the present study, the symmetric SO2stretching vibrations were obtained at 1 127 and 1 092 cm-1in FTIR spectra. The bands observed 1 082 cm-1in FT-Raman spectra were assigned to antisymmetric SO2stretching vibrations. A major coincidence of experimental values with that of literature[38-39]and theoretical results are found for above conclusions. The bending vibrations of O—S—O are also given in Table 1.
3.7 Ring vibrations
The ring stretching vibrations are very much important in the spectrum of aromatic compounds and are highly characteristic of the aromatic ring itself. However, empirical assignments of vibrational modes for peaks in the fingerprint region are difficult. Bands between 1 400 and 1 650 cm-1in benzene derivatives are assigned to ring vibrations. In general, the bands are of variable intensity and observed at 1 625~1 590, 1 590~1 575, 1 540~1 470, 1 460~1 430 and 1 380~1 280 cm-1from the frequency ranges given by Varsanyi[40]for the five bands in the fingerprint region. In the present work, the bands with are of different intensity and observed at 1 490, 1 332, 1 222, 1 195, 1 160, 1 153, 1 127 and 520 cm-1in FTIR and 1 530, 1 337, 1 248, 1 225, 1 154, 615 aand 518 cm-1in FT-Raman have been assigned to C—C stretching vibrations. The theoretically calculated values at 1 601, 1 631, 1 492, 1 415, 1 330, 1 307, 1 305, 1 253, 1 229, 1 199, 1 198, 1 164, 1 161 and 621 cm-1by B3LYP/6-31G(d,p) method shows excellent agreement with experimental data. The C—C in-plane and out-of-plane bending vibrations are the modes associated with smaller force constants than the stretching ones, and hence assigned to lower frequencies. The in plane deformations are at higher frequencies than the out-of-plane vibrations. Shimanouchi et al.[41]gave the frequency data for these vibrations for different benzene derivatives as a result of normal coordinate analysis. Although some modes are missing in experimental spectra, however, the calculated CCC in-plane and out-of-plane bending modes are found to be consistent with the recorded spectral values, as can be seen in Table 2.
4 Conclusion
FTIR, FT-Raman and DFT quantum chemical calculations studies were performed on HPAEPMS, in order to identify their structural and vibrational features. Several properties were carried out using experimental techniques and tools derived from density functional theory. On the basis of experimental results and TED calculations, assignments of all the fundamental vibrational frequencies were done. A good correlation between the observed and scaled wave number was obtained for the title compound. Scaled results seemed to be in good agreement with experimental ones. We hope our results will be of assistance in the quest of the experimental and theoretical evidence for the HPAEPMS molecule in reaction intermediates, nonlinear optical and photoelectric materials and will also be helpful for the design and synthesis of new materials.
[1] Maren T H. Ann. Rev. Pharmacol. Toxicol., 1976, 16: 309.
[2] Supuran C T, Scozzafava A. Curr. Med. Chem. Immunol. Endocrine Metabolic Agents 1, 2001. 61.
[3] Supuran C T, Scozzafava A, Casini A. Med. Res. Rev., 2003, 23: 146.
[4] Supuran C T, Scozzafava A. Expert Opin. Ther. Patents, 2002, 12: 217.
[5] Silverman R B. The Organic Chemistry of Drug Design and Drug Action, Academic, London, 1992.
[6] Albala D M, Prien E L, Galal H A. J. Endourol., 1994, 8: 401.
[7] Reynolds J E F(Ed.). Martindale: The Extra Pharmacopoeia, 31st ed., The Royal Pharmaceutical Society, London, 1996.
[8] Kaur I P, Singh M, Kanwar M. Int. J. Pharm. 2000, 199: 119.
[9] Famaey J P. Inflamm. Res., 1997, 46: 437.
[10] OnucCozar, LászlóSzabó, Vasile Chiʂ , et al. Annals of the Academy of Romanian Scientists Physics Series, 2010, 2: 17.
[11] Nagabalasubramanian P B, Periandy S. Spectrochim. Acta A, 2010, 77: 1099.
[12] Govindarajan M, M. Karabacak, A. Suvitha, S. Periandy, Spectrochim. Acta A, 2012, 89: 137.
[13] Govindarajan M, Karabacak M, Periandy S, et al. Spectrochim. Acta A, 2012, 97: 231.
[14] Karabacak M, Kurt M, Atac A. J. Phys. Org. Chem., 2009, 22: 321.
[15] Mehmet Karabacak, Dilek Karagöz, Mustafa Kurt. Spectrochim. Acta A, 2009, 72: 1076.
[16] Palafox M A, Tardajos G, Martines A G, et al. Chem. Phys., 2007, 340: 17.
[17] Stephens P J, Devlin F J, Chavalowski C F, et al. J. Phys. Chem., 1994, 98: 11623.
[18] Devlin F J, Finley J W, Stephens P J, et al. J. Phys. Chem., 1995, 99: 16883.
[19] Lee S Y, Boo B H. Bull. Korean Chem. Soc., 1996, 17: 754.
[20] Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 03, Revision A.I, Gaussian, Inc., Pittsburgh, PA, 2003.
[21] Schlegel H B. J. Comput. Chem., 1982, 3: 214.
[22] Foresman J B, Frisch A. Exploring Chemistry with Electronic Structure Methods,Gaussian Inc., Pittsburgh, 1996.
[23] Michalska D. Raint Program, Wroclaw University of Technology, 2003.
[24] Michalska D, Wyokinski R. Chem. Phys. Lett., 2005, 403: 211.
[25] Sanchez-Camazano M, Sanchez-Martin M J, Vicente M T, et al. Clay Minerals, 1987, 22: 121.
[26] Silverstein M, Bassler G C, Morril C. Spectroscopic Identification of Organic Compounds, John Wiley, Newyork, 1981.
[27] Bellamy L J. The Infrared Spectra of Complex Molecules, Vol. 2, Chapman and Hall, London, 1980.
[28] Chandran A, Mary Y S, Varghese H T, et al. Spectrochim. Acta A, 2011, 79: 1584.
[29] Rastogi V K, Palafox M A, Tanwar R P, et al. Spectrochim. Acta, 2002, 58A: 1987.
[30] Roges N P G. A Guide to the Complete Interpretation of the Infrared Spectra of Organic Structures, Wiley, New York, 1994.
[31] Kalsi P S. Spectroscopy of Organic Compounds, Wiley Eastern Limited, New Delhi, 1993.
[32] Sajan D, Joe I H, Jayakumar V S. J. Raman Spectrosc., 2005, 37: 508.
[33] Reddy B V, Rao G R. Vib. Spectrosc., 1994, 6: 231.
[34] Areanas J F, Tocn I L, Otero J C, et al. J. Mol. Struct., 1997, 410: 443.
[35] Sebastian S, Sundaraganesan N. Spectrochim. Acta A, 2010, 75: 941.
[36] Sekino H, Bartlett R J. J. Chem. Phys., 1986, 84: 2726.
[37] Henriksson J, Saue T, Norman P. J. Chem. Phys., 2008, 128: 105.
[38] Sun Y X, Hao Q L, Yu Z X, et al. Mol. Phys., 2009, 107: 217.
[39] Ahmed A B, Feki H, Abid Y, et al. J. Mol. Struct., 2009, 920: 1.
[40] Varsanyi G. Assignments for Vibrational Spectra of Seven Hundred Benzene Derivatives, Vol. 1/2, Academic Kiado, Budapset, 1973.
[41] Shimanouchi T, Kakiuti Y, Gamo I. J. Chem. Phys., 1956, 25: 1245.
O657.3
A
2015-08-29; accepted: 2016-01-11
10.3964/j.issn.1000-0593(2016)03-0880-07
*Corresponding author email: n_rishibalaa@yahoo.co.in
