氧化应激促进AGT-REN双转基因高血压小鼠心肌中电导钙激活钾离子通道表达*
2016-06-06杨会笃苏叶馨赵宏跃王丽萍
周 骁, 杨会笃, 苏叶馨, 赵宏跃, 王丽萍
(华北理工大学基础医学院生理学系,河北 唐山 063000)
氧化应激促进AGT-REN双转基因高血压小鼠心肌中电导钙激活钾离子通道表达*
周骁,杨会笃,苏叶馨,赵宏跃,王丽萍△
(华北理工大学基础医学院生理学系,河北 唐山 063000)
[摘要]目的: 探讨心肌重构过程中心肌中电导钙激活钾离子通道(KCa3.1)蛋白表达与氧化应激反应之间的关系。方法: 雄性血管紧张素-肾素(AGT-REN)双转基因高血压(dTH)小鼠2、4、8、12月龄各6只进行相应指标检测。另取12只6月龄雄性dTH小鼠,随机分为2组:模型组(dTH组)和N-乙酰半胱氨酸(NAC)组。同时取6只dTH同品系野生C57B6小鼠作为对照(WT组)。NAC组小鼠腹腔注射NAC 400 mg·kg-1·d-1,WT和dTH组小鼠腹腔注射等量生理盐水。4周后检测各组小鼠血压变化;ELISA法测定小鼠血浆中AngⅡ和Ang(1-7)含量变化;试剂盒检测心肌组织超氧化物歧化酶(SOD)活性及丙二醛(MDA)含量;Western blot法检测小鼠心肌胶原蛋白collagen I、collagen III及KCa3.1蛋白表达的变化。结果: dTH组小鼠血压、血浆AngⅡ、MDA及胶原蛋白含量均高于同龄WT组小鼠(2月龄除外),并随年龄增长而增高(P<0.05);血浆Ang(1-7)和心肌SOD活性随年龄增长下降,并低于同龄WT组小鼠(2月龄除外)(P<0.05)。NAC干预使6月龄dTH小鼠心肌SOD活性增强(P<0.01),同时心肌MDA、胶原蛋白和KCa3.1蛋白表达下降(P<0.05)。结论: 高血压所致心肌重构过程中心肌KCa3.1蛋白表达增加可能与心肌氧化应激水平增强有关。
[关键词]心肌重构; 氧化应激; KCa3.1
心肌重构是多种心脏疾病中的重要病理过程,最终发展为心力衰竭。血管紧张素II(angiotensin II, Ang II)可诱导心肌成纤维细胞分化、增殖及合成胶原,降低基质金属蛋白酶活性,是心肌重构发生的主要诱导因素。Ang II还可通过激活氧化应激反应而诱发心肌活性氧(reactive oxygen species,ROS)生成增多。研究显示,ROS可通过多种与心肌肥大有关的信号激酶和转录因子,诱导机械拉伸或神经激素性刺激诱发的心肌肥厚性反应,进而在心肌重构的发生过程中发挥重要作用[1-3]。
中电导钙激活的钾通道(intermediate conduc-tance Ca2+-activated K+channel, KCa3.1)的作用主要是通过控制膜电位调节进入细胞内的钙进而调节钙信号。KCa3.1介导的细胞内钙升高是许多细胞增殖所必需的。研究发现,Ang II和晚期糖基化终产物(advanced glycation end products,AGEs)通过与其受体结合,分别激活ERK1/2、p38 MAPK、PI3K等信号转导通路,诱导KCa3.1活性及表达上调,促进大鼠心肌成纤维细胞增殖及胶原合成[4-5]。对KCa3.1的研究还显示,使用特异性阻断剂阻断KCa3.1可减轻压力超负荷高血压大鼠心肌组织的炎症反应[6]。这些结果表明,KCa3.1与心肌重构关系密切,可能是多种因素诱导心肌重构发生的一个重要靶点。
血管紧张素-肾素(angiotensin-renin,AGT-REN)双转基因高血压小鼠 (double transgenic hypertension mice,dTH)是在野生C57B6小鼠基础上采用分子生物学方法人为转入人肾素(hRen+/+)和血管紧张素(hAgt+/+)的基因小鼠。我们前期研究显示,dTH小鼠具有血压增高及继发的一系列病理表现。本研究通过对不同年龄高血压小鼠各项指标的检测及进行药物干预,探讨心肌重构过程中KCa3.1通道蛋白表达与氧化应激反应之间的关系,结果将进一步明确KCa3.1通道在心肌重构中的重要作用。
材料和方法
1实验动物和分组
2~12月龄雄性dTH小鼠,体重22~30 g,由华北理工大学生理教研室提供。
6月龄雄性dTH小鼠随机分为:模型组(dTH组);N-乙酰半胱氨酸(N-acetylcysteine,NAC)组;同时取6只dTH同品系野生型(wild-type,WT)C57B6小鼠作为对照。NAC组小鼠腹腔注射NAC(400 mg·kg-1·d-1),WT和模型组小鼠腹腔注射等量生理盐水。4周后进行各项指标的测定。
2主要试剂
NAC购自Sigma;Western blot 相关试剂购自 Amresco;超氧化物歧化酶(superoxide dismutase,SOD)和丙二醛(malondialdehyde,MDA)试剂盒购自南京建成生物工程研究所;兔抗collagen I和collagen III多克隆抗体购自博士德;兔抗KCa3.1多克隆抗体以及Ang II、Ang (1-7) ELISA试剂盒购自Abcam。
3 实验方法
3.1血压的测定无创动脉血压仪(北京软隆科技有限责任公司)测量各组动物血压。血压仪主机与电脑相连,保温器温度维持在37 ℃。将小鼠尾根部放入加压感应器中,当血压计感应到适当的鼠尾血流状态时,开始自动测量,所测数值和感应波形自动存储于电脑。
3.2ELISA法测定血浆中AngⅡ和Ang(1-7)含量小鼠眼球取血,4 ℃放置过夜后,5 000 r/min离心取上清,按照AngⅡ和Ang(1-7)ELISA试剂盒说明书进行测定。
3.3心肌组织SOD活性及MDA含量检测取100 mg心肌组织置于1 mL预冷PBS中匀浆,1 500 r/min离心取上清。按照SOD试剂盒及MDA试剂盒说明书操作。
3.4Western blot法检测各组小鼠心肌collagen I、collagen III和KCa3.1蛋白表达的变化100 mg心肌组织裂解提取总蛋白,BCA法测蛋白总浓度。取等量蛋白于10% SDS-PAGE分离,90 V转膜1 h将蛋白转至PVDF膜。5% 奶粉封闭1 h后,滴加 I 抗anti-collagen I(1∶500)、anti-collagen III(1∶500)和anti-KCa3.1(1∶300),4 ℃过夜。滴加 II 抗,室温孵育1 h,化学发光试剂放射自显影。
4统计学处理
采用SPSS 16.0统计软件,计量数据以均数±标准差(mean±SD)表示,组间比较用单因素方差分析,两两比较使用Bonferroni检验。采用SigmaPlot 10.0软件做统计图。以P<0.05为差异有统计学意义。
结果
1血浆中Ang Ⅱ和Ang(1-7)含量的变化
ELISA结果显示,随着年龄增长,dTH小鼠循环血液中Ang Ⅱ含量逐渐升高,除2月龄外,均高于同龄WT组小鼠(P<0.05);而Ang(1-7)含量则呈下降趋势,并低于同龄WT组小鼠(P<0.05),见图1。
2血压的测定
对2、4、6、8、12月龄dTH小鼠的血压进行检测,结果发现,随着年龄增长,dTH小鼠平均动脉血压(mean arterial blood pressure,MAP)逐渐升高,除2月龄组外均显著高于同龄野生型小鼠(P<0.01),见图2A。6月龄dTH小鼠给予NAC治疗后,MAP出现明显下降(P<0.01),见图2B。
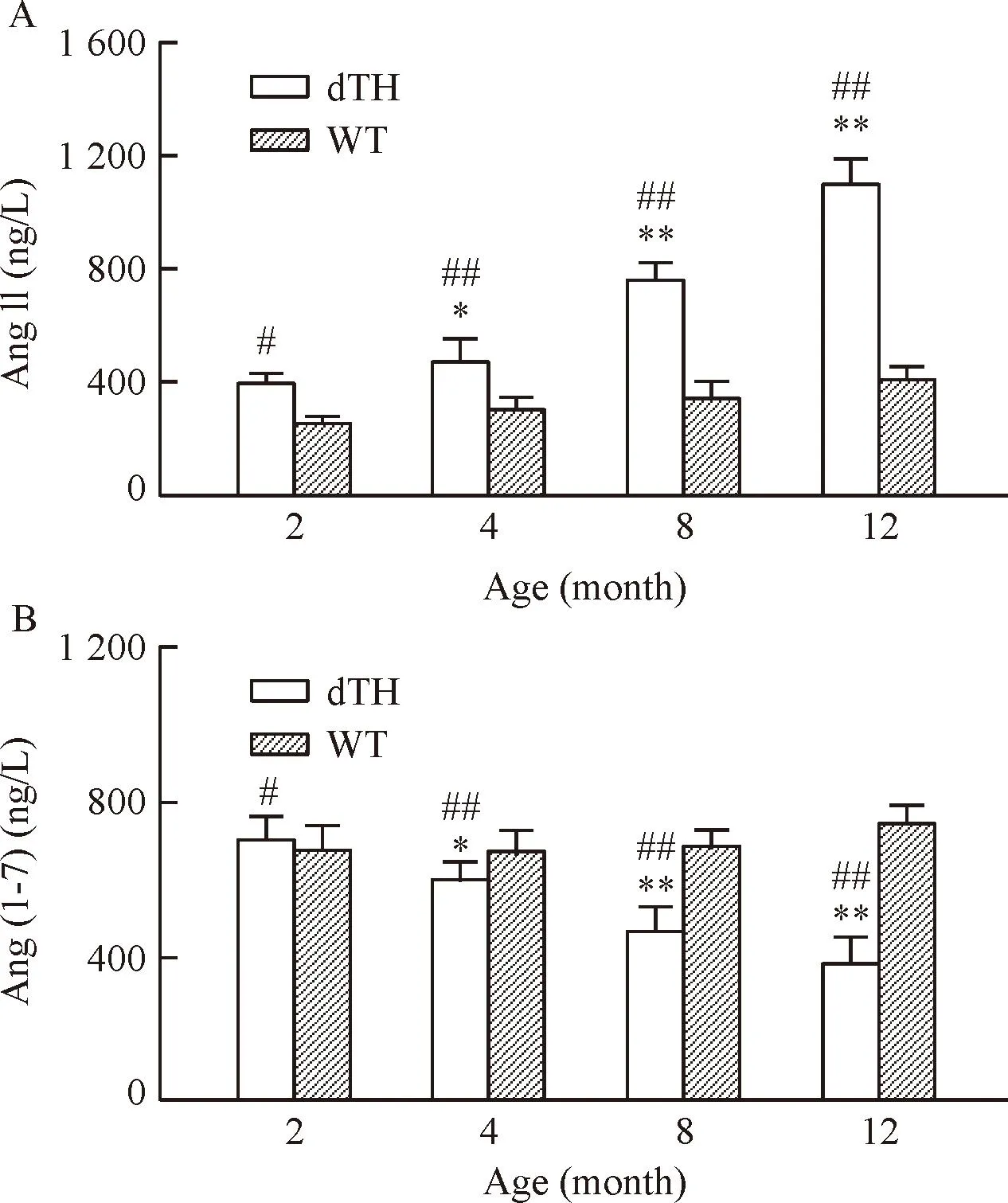
Figure 1.Concentrations of Ang II (A) and Ang (1-7) (B) in the plasma of mice. Mean±SD.n=6.#P<0.05,##P<0.01 vs WT;*P<0.05,**P<0.01 vs 2 months.
图1小鼠血浆Ang II和Ang (1-7)含量的变化

Figure 2.MAP of mice. A: MAP of the mice at different ages; B: MAP of 6-month-old dTH mice treated with intra-peritoneal injection of vehicle or NAC (400 mg·kg-1·d-1) for 4 weeks. Mean±SD.n=6.**P<0.01 vs WT;#P<0.05,##P<0.01 vs 2 months;△△P<0.01 vs dTH.
图2小鼠血压的变化
3心肌组织SOD活性及MDA含量的变化
如图3所示,随着年龄增长,dTH小鼠心肌的MDA含量逐渐升高,其中,4、8、12月龄dTH小鼠MDA含量均明显高于同龄野生型小鼠(P<0.01);而SOD活性随着年龄增长逐渐下降,并低于同龄野生型小鼠(P<0.05)。6月龄dTH小鼠使用抗氧化剂NAC干预1个月后,与同月龄dTH小鼠相比,心肌MDA含量下降(P<0.01),SOD活性升高(P<0.01)。
4心肌胶原蛋白表达变化
dTH小鼠心肌collagen I和collagen III表达随年龄增长逐渐升高(P<0.05)。6月龄高血压小鼠使用抗氧化剂NAC干预 1个月后,与同月龄高血压组小鼠相比,心肌组织中collagen I和collagen III表达明显下降(P<0.01),见图4。
5心肌KCa3.1蛋白表达变化
dTH小鼠心肌KCa3.1蛋白表达随年龄增长逐渐升高(P<0.05),其中,4、8、12月龄dTH小鼠KCa3.1蛋白表达均明显高于同龄野生型小鼠(P<0.01),与MDA含量变化趋势一致。6月龄dTH小鼠使用抗氧化剂NAC干预 1 个月后,与同月龄dTH组小鼠相比,心肌组织中KCa3.1蛋白表达明显下降(P<0.01),见图5。
讨论
心肌重构是心肌在长期超负荷、神经体液因素及其它促生长因子影响下,出现的心肌细胞肥大、细胞外基质增加、心肌纤维化等形态学改变,同时伴有心脏功能的减退。Ang II诱发的氧化应激反应是心肌重构发生的重要因素。氧自由基的产生和消除失衡,或外源性氧化物质的过量摄入使ROS在体内或细胞内过量蓄积,而ROS可直接刺激心肌成纤维细胞增殖,同时激活基质金属蛋白酶,导致细胞外基质重塑。ROS还参与多种因素诱发的心肌肥厚反应,并由此引发心血管疾病[3, 7-8]。
dTH小鼠是一种特征稳定的较好的高血压动物模型。对不同月龄的dTH小鼠的研究结果显示,随着年龄增长,高血压小鼠循环血液中Ang II水平逐渐增高,与之相对的,Ang(1-7)的含量随年龄增长而下降。同时,小鼠心肌组织中脂质过氧化物MDA表达随年龄增长而上升,SOD活性则逐渐下降。研究证实,Ang(1-7)在心脏重塑过程中发挥着重要负向调节作用,是对抗Ang II生物效应的主要作用机制[9]。这一结果提示,高血压小鼠心肌中氧化应激水平随着Ang II浓度的的升高而增强,而Ang(1-7)水平下降。
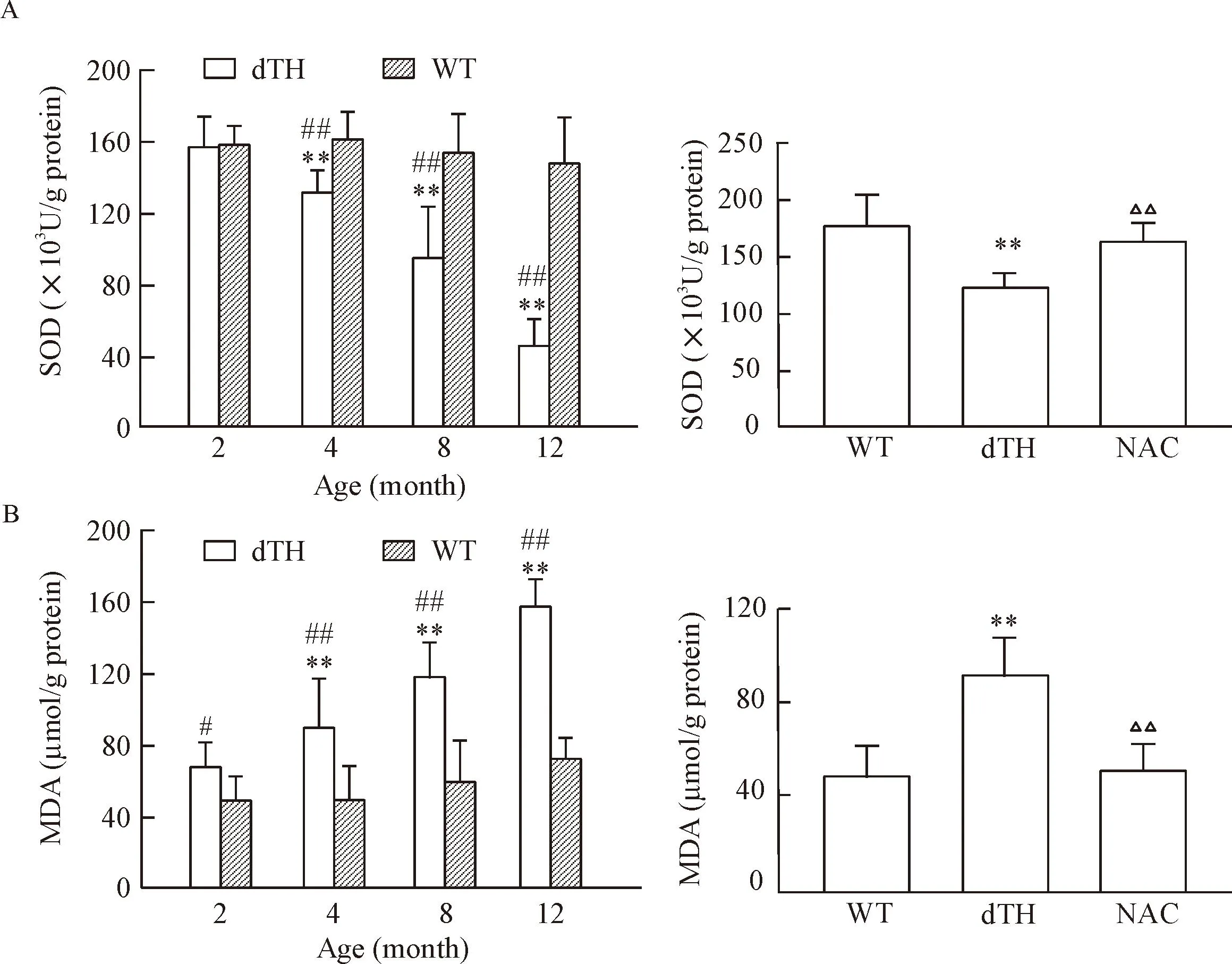
Figure 3. The activity of SOD (A) and content of MDA (B) in the myocardium of the mice. Mean±SD.n=6.*P<0.05,**P<0.01 vs WT;#P<0.05,##P<0.01 vs 2 months;△△P<0.01 vs dTH.
图3AGT-REN高血压小鼠心肌SOD活性及MDA含量的变化
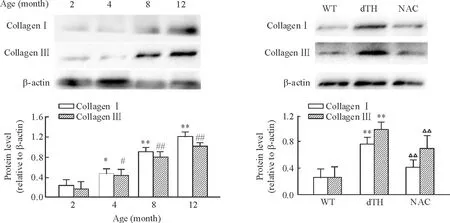
Figure 4.The protein expression of collagen I and collagen III in the myocardium of AGT-REN mice. Mean±SD.n=6.*P<0.05,**P<0.01 vs WT;#P<0.05,##P<0.01 vs 2 months;△△P<0.01 vs dTH.
图4AGT-REN高血压小鼠心肌胶原蛋白表达变化
KCa3.1在成纤维细胞增殖中发挥作用早有报道。研究显示,KCa3.1基因敲除或用TRAM-34药理学阻断其作用可大大降低小鼠和大鼠由单侧后输尿管梗阻诱发的肾纤维化[10]。阻断糖尿病小鼠KCa3.1可降低由转化生长因子β1(transforming growth factor-β1,TGF-β1)诱导的肾成纤维细胞中I型胶原、纤连蛋白、肌动蛋白、波形蛋白及纤维蛋白-1的表达[11];在敲除KCa3.1基因的模型小鼠也得到相同结果[12]。另外,靶向阻断KCa3.1可降低由TGF-β1诱导的大鼠肾小球系膜细胞的老化、向肌成纤维细胞的分化及增殖[13],进而减轻了肾小球的硬化。对心脏的研究显示,KCa3.1在梗死后的大鼠心肌血管细胞表达增强,并出现在缺血区域的单核细胞和心肌成纤维细胞上。本研究中,我们检测了不同月龄dTH小鼠心肌KCa3.1蛋白的表达,结果显示,随着年龄增长,小鼠心肌中KCa3.1蛋白表达逐渐增加,与氧化应激反应增强趋势一致,提示dTH小鼠心肌KCa3.1蛋白表达的增加可能与心肌氧化应激水平增高有关。NAC是细胞内还原型谷胱甘肽的前体,具有干扰自由基生成、清除已生成的自由基、抗氧化等作用,为明确心肌KCa3.1表达与氧化应激水平的关系,我们选取6月龄高血压小鼠,给予ROS清除剂NAC进行干预,结果显示,NAC在增强高血压小鼠SOD活性,降低血压及心肌MDA、胶原蛋白表达的同时,抑制了高血压小鼠心肌组织KCa3.1蛋白的表达。这一结果进一步表明,dTH小鼠心肌KCa3.1蛋白表达的增多与氧化应激反应的增强有关。
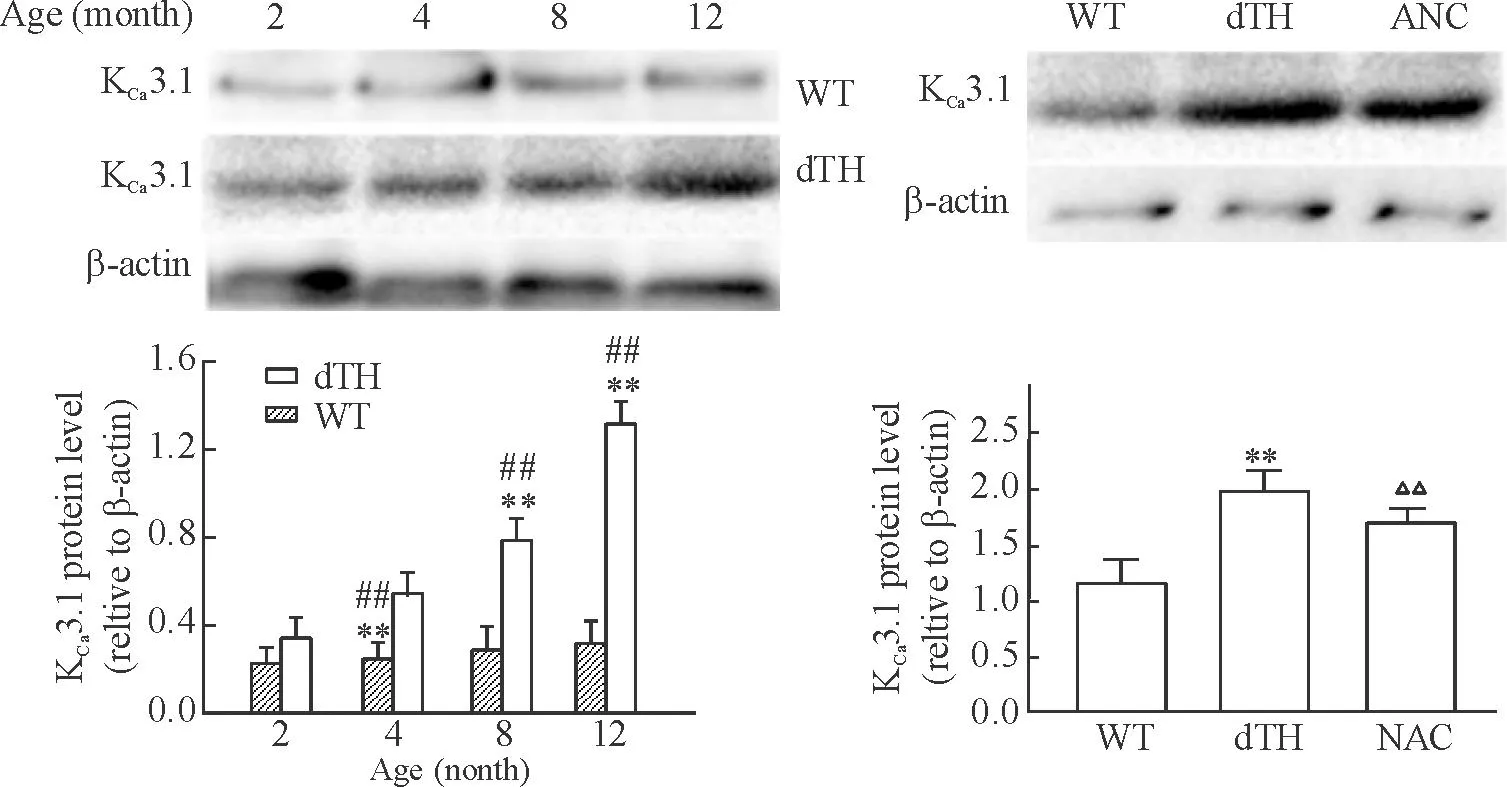
Figure 5.The protein expression of KCa3.1 in the myocardium of mice. Mean±SD.n=6.**P<0.01 vs WT;##P<0.01 vs 2 months;△△P<0.01 vs dTH.
图5小鼠心肌KCa3.1蛋白表达的变化
[参考文献]
[1]Santos CX, Anilkumar N, Zhang M, et al. Redox signaling in cardiac myocytes[J]. Free Radic Biol Med, 2011, 50(7):777-793.
[2]Touyz RM, Briones AM. Reactive oxygen species and vascular biology: implications in human hypertension[J]. Hypertens Res, 2011, 34(1): 5-14.
[3]Ward NC,Croft KD.Hypertension and oxidative stress[J].Clin Exp Pharmacol Physiol, 2006, 33(9):872-876.
[4]Wang LP, Wang Y, Zhao LM, et al. Angiotensin II upregulates KCa3.1 channels and stimulates cell proliferation in rat cardiac fibroblasts[J]. Biochem Pharmacol, 2013, 85(10):1486-1494.
[5]Zhao LM, Zhang W, Wang LP, et al. Advanced glycation end products promote proliferation of cardiac fibroblasts by upregulation of KCa3.1 channels[J]. Pflugers Arch, 2012, 464(6):613-621.
[6]Zhao LM, Wang LP, Wang HF, et al. The role of KCa3.1 channels in cardiac fibrosis induced by pressure overload in rats[J]. Pflugers Arch, 2015, 467(11):2275-2285.
[7]Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure[J]. Am J Physiol Heart Circ Physiol, 2011, 301(6):H2181-H2190.
[8]Papaharalambus CA, Griendling KK. Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury[J]. Trends Cardiovasc Med, 2007, 17(2):48-54.
[9]梁伟杰, 陈景福, 宋明才,等. 血管紧张素-(1-7)/Mas受体轴通过调控NF-κB通路保护心肌细胞对抗高糖诱导的损伤[J]. 中国病理生理杂志, 2015, 31(2): 267-273.
[10]Albaqumi M, Srivastava S, Li Z, et al. KCa3.1 potassium channels are critical for cAMP dependent chloride secretion and cyst growth in autosomal-dominant polycystic kidney disease[J]. Kidney Int, 2008, 74(6):740-749.
[11]Huang C, Shen S, Ma Q, et al. KCa3.1 mediates activation of fibroblasts in diabetic renal interstitial fibrosis[J]. Nephrol Dial Transplant, 2014, 29(2):313-324.
[12]Huang C, Shen S, Ma Q, et al. Blockade of KCa3.1 ameliorates renal fibrosis through the TGF-β1/Smad pathway in diabetic mice[J]. Diabetes, 2013, 62(8):2923-2934.
[13]Fu RG, Zhang T, Wang L, et al. Inhibition of the K+channel KCa3.1 reduces TGF-β1-induced premature senescence, myofibroblast phenotype transition and proliferation of mesangial cells[J]. PLoS One, 2014, 9(1):e87410.
(责任编辑: 林白霜, 罗森)
Oxidative stress promotes KCa3.1 expression in myocardium of AGT-REN double transgenic hypertension mice
ZHOU Xiao, YANG Hui-du, SU Ye-xin, ZHAO Hong-yue, WANG Li-ping
(Department of Physiology, School of Basic Medicine, North China University of Science and Technology, Tangshan 063000, China. E-mail: zip_2000@163.com)
[ABSTRACT]AIM: To study the relationship between oxidative stress reaction and KCa3.1 protein expression in the process of myocardial remodeling.METHODS: The male AGT-REN double transgenic hypertension (dTH) mice (2, 4, 8 and 12 months old, each n=6) were used to detect the changes of the corresponding indexes. The male dTH mice (6 months old) were randomly divided into 2 groups: dTH group and N-acetylcysteine (NAC) group, and 6 wild-type(WT) C57B6 mice served as controls. The mice in NAC group were treated with NAC at dose of 400 mg·kg-1·d-1, and the WT and model mice were treated with normal saline. After 4 weeks, the concentrations of Ang Ⅱ and Ang (1-7) in the plasma were measured by ELISA. The superoxide dismutase (SOD) activity and malondialdehyde (MDA) content were detected using SOD and MDA kits. The protein levels of collagen I, collagen III and KCa3.1 were determined by Western blot. RESULTS: Compared with the WT mice, mean arterial blood pressure (MAP), concentration of Ang Ⅱ in the plasma, the content of MDA, and the protein expression of collagens and KCa3.1 in the myocardium of 4-, 8- and 12-month-old dTH mice were increased, and gradually raised along with the age from 2 to 12 months (P<0.05), but the SOD activity in myocardium and Ang (1-7) content in plasma were decreased (P<0.01). NAC reduced the MAP, the content of MDA, and the protein expression of collagens and KCa3.1 in the myocardium of 6-month-old dTH mice (P<0.05 or P<0.01), and increased SOD activity compared with dTH group (P<0.01).CONCLUSION: Increased protein expression of myocardial KCa3.1 during myocardial remodeling induced by hypertension may be associated with enhancement of myocardial oxidative stress level.
[KEY WORDS]Myocardial remodeling; Oxidative stress; KCa3.1
[文章编号]1000- 4718(2016)05- 0869- 05
[收稿日期]2015- 10- 26[修回日期] 2016- 03- 11
*[基金项目]河北省自然科学基金资助项目(No. H2015209153); 华北理工大学大学生创新创业训练计划(No. X2015145)
通讯作者△Tel: 0315-3725606; E-mail: zip_2000@163.com
[中图分类号]R542.2;R363
[文献标志码]A
doi:10.3969/j.issn.1000- 4718.2016.05.017
杂志网址: http://www.cjpp.net
