Ne
—Na2分子基态势能面的从头算及分析拟合
2016-05-25李伟艳汪小润
王 悦, 李伟艳, 汪小润, 张 飞, 张 强, 方 芳
(1.铜陵学院电气工程学院,铜陵 244000; 2.铜陵市环保局,铜陵 244000)
Ne
—Na2分子基态势能面的从头算及分析拟合
王悦1*, 李伟艳1, 汪小润1, 张飞1, 张强1, 方芳2
(1.铜陵学院电气工程学院,铜陵 244000; 2.铜陵市环保局,铜陵 244000)
摘要:采用aug-cc-pVQZ(弥散函数的基组)/Na、aug-cc-pVDZ(弥散函数的基组)/Ne以及中点键函数的大基组,使用单、双迭代并包含三重激发微扰校正的耦合簇CCSD(T)理论方法,计算了Ne—Na2基态146个基态单点能.通过拟合96个参数,给出了Ne—Na2三原子分子体系的基态分子势能函数的解析表达式,并分析了其基态二维势能面的特性,在此基础上绘出了Ne—Na2三原子分子体系的三维势能曲线.计算结果表明,Ne—Na2基态势能面存在2个较浅的势阱,对应于θ=80°,RNe—Na=3.3a0处,势阱的阱深约为-6.750×10-1cm-1和线型结构θ=0°,RNe—Na=10a0处,势阱深度约为-2.341×10-3cm-1.此三原子分子体系的势能面呈现出弱的角度各向异性.
关键词:Ne—Na2; CCSD(T); 分子基态; 势能面; 从头算
研究冷分子反应和碰撞具有重要意义[1-2].一方面,超冷分子物理的发展为波色-爱因斯坦凝聚体、广义相对论时空特性的探索、费米简并气体量子相变的精密测量及量子计算机的发展等开辟了新的路径.另一方面,用冷原子缓冲气载带冷却[3-4]、光缔合[5-6]、磁/电场调节引发连续-束缚态Feshbach共振[7-8]等方法可实现碱金属双原子分子(异核或同核) 的冷却或囚禁,因此,研究Na2的相关特性,对于冷分子物理的研究和应用具有重要价值.
在单体刚性椭球模型基础上,引入多体刚性椭球模型,成功地解决了单体刚性椭球模型在处理碱金属分子与稀有气体原子碰撞时的局限性[9].臧华平等[10]用多体刚性椭球模型研究了Ne—Na2碰撞体系的振动转动激发速率常数,并计算了相对入射能量为Er=190 meV时,氖同位素原子20Ne、34Ne与钠的同位素分子18Na2、23Na2、37Na2的碰撞态转动激发积分散射截面和总转动激发积分散射截面,讨论钠同位素替代对转动激发积分散射的影响,总结出20Ne、34Ne—18Na2、23Na2及37Na2碰撞体系转动激发积分散射截面随着Na分子质量变化的规律,在此基础上计算了相互作用势的不同区域对20Ne—18Na2、23Na2及37Na2碰撞体系转动激发积分散射截面的贡献情况,分析了相互作用势的不同区域对转动激发积分散射截面的影响.BERGMANN等[11]近似计算研究了积分和微分散射截面,提出了Ne—Na2体系的势能面.但上述研究均不涉及Ne—Na2体系的多项式展开势能函数表达式.目前,有关Ne—Na2分子的精确势能面的研究报道尚少.
本文采用大的基组和高级别从头算方法,研究Ne—Na2体系的相互作用势.得到了更精确的三维势能面,对Ne—Na2体系理论和实验提供了可靠的依据.
1物理模型和方法
1.1模型选择
由于Ne—Na2体系属于三原子分子,将把Na—Na固定在x轴,采用Jacobi坐标系(r,R,θ).其中,Ne原子与Na—Na中心连线夹角为θ,如图1所示,其中r表示Na—Na间距,中点为坐标原点,R表示原点到Ne原子距离,为了使计算中R的取值合理,即实际中Ne原子与Na—Na不能靠得太近,选取了键函数Bq,并放置在原点到Ne原子的连线中点.
基态时req=3.079a0,那么在1个给定的θ,变换R(从1a0到 20a0,取步长为1a0,其中在非势阱区域取点稀疏,而接近势阱区域取点密集),再变换角度θ(从0°变化步长为10°).预算时发现在θ=80°和θ=0°出现势阱,因此在短程区1a0≤R≤2a0计算时取ΔR=0.1a0;在势阱附近2a0≤R≤3a0,取ΔR=0.05a0;在长程弱相互作用区域即4a0≤R≤20a0取ΔR=1a0,共计算了146个几何构型.所有计算在G09程序包[12]中完成.

图1 Ne—Na2体系计算坐标
1.2计算方法
为了选择合理的基组计算势能面,在靠近势阱区的θ=90°处,计算了Ne取几种不同基组的势能曲线(图2),几种基组在长程区域较吻合,在势阱区和短程区域出现误差.计算中,选取短程和势阱区相对误差都较小的aug-cc-pVQZ /Na、aug-cc-pVDZ/Ne[13]作为基组,进行势能面的扫描.
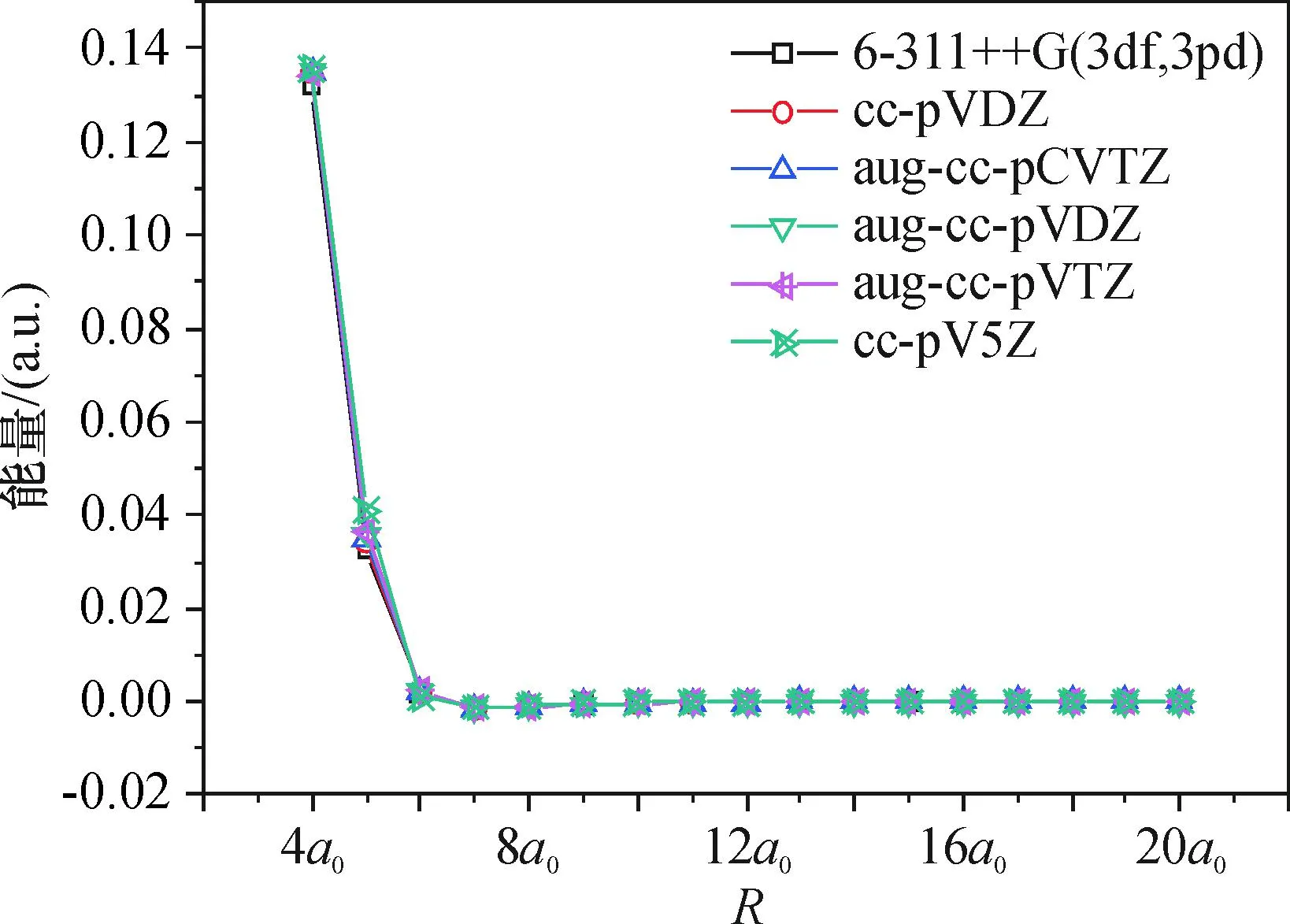
图2 Ne—Na2体系在r=re、θ=90°时不同基组的能量
采用FCP(Full Couterpoise)方法[14]消除基函数的重叠误差.在超分子近似中,FCP方法要求用整个体系的基组在相应的几何构型下分别计算聚合体及各单体的能量,相互作用能则为相应能量的差值:
V(R,θ)=ESi-H2[Si+H-H]-
ESi[Si+H-H]-EH2[Si+H-H],
(1)
为了便于碰撞动力学研究,将从头算得到的势能值用最小二乘法拟合成1个解析势能函数:
V(q,R,θ)=Vsh(q,R,θ)+Vas(q,R,θ),
(2)
其中,Vsh(q,R,θ)和Vas(q,R,θ)分别表示相互作用的短程部分和长程部分[15].短程部分的函数形式为
Vsh(q,R,θ)=G(q,R,θ)e[D(q,θ)-B(q,θ)R],
(3)

(4)

(5)
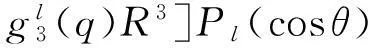
(6)


(7)

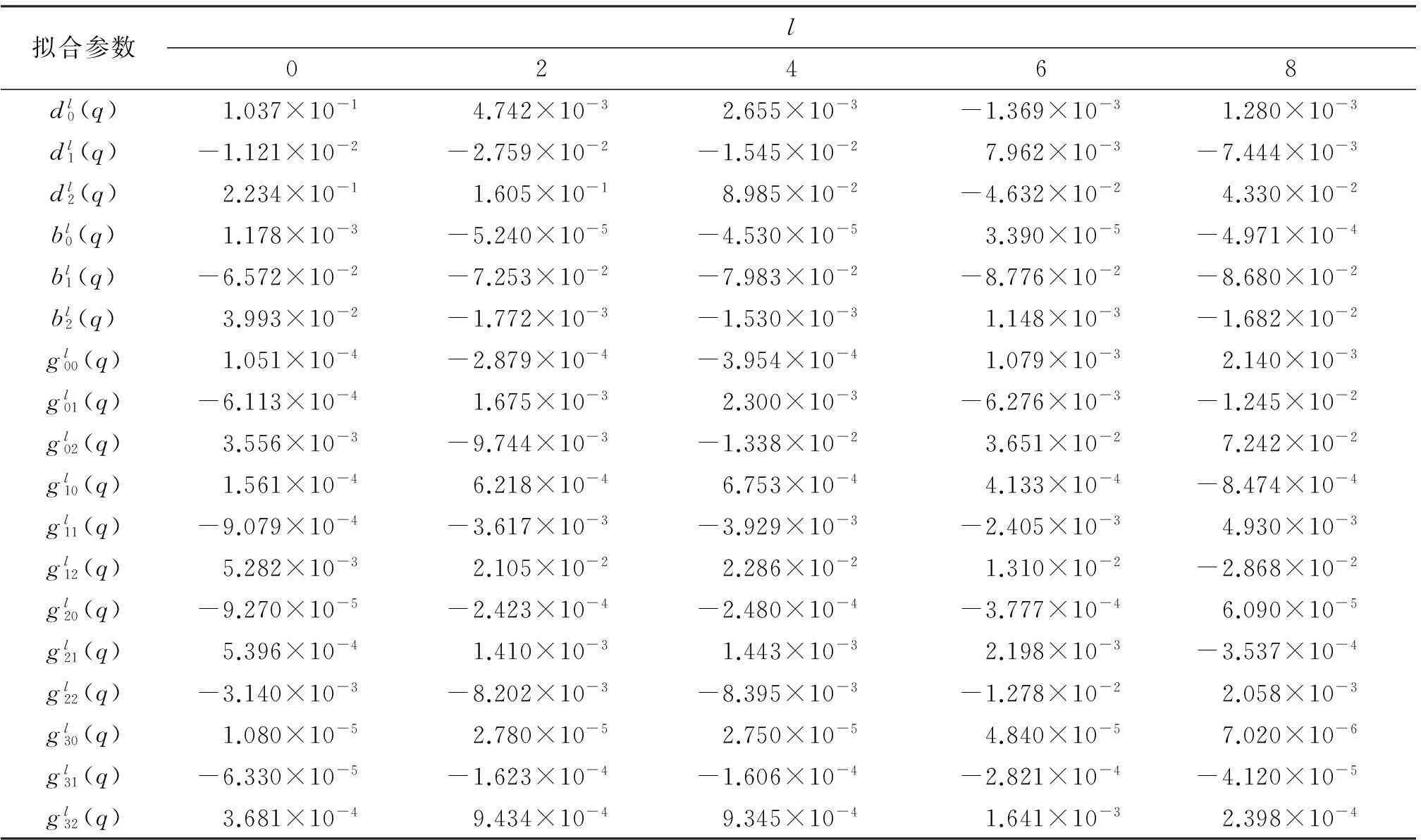
表1 Ne—Na2体系相互作用解析势能函数拟合参数
2结果与讨论
从不同角度描绘出势阱位置(图3).长程7a0之后才出现明显的吸引,势能面上有1个极小值,对应于θ=80°,RNe—Na=3.3a0处,阱深约为-6.750×10-1cm-1;线型结构θ=0°,RNe—Na=10a0处,阱深约为-2.341×10-3cm-1.可以看出Ne—Na2体系势能面上势阱非常浅,说明Ne—Na2体系相互作用势较弱.
化学键中,原子基本保持一定的距离,其中,势能阱是材料物理性质的一个重要数据.化合物中的原子结合是否紧密,将直接影响熔点、弹性模量和热膨胀系数等性质.较深的势能阱表示原子间结合较紧密,其对应的材料较难熔融,并具有较高的弹性模量和较低的热膨胀系数.经拟合后画出平衡位置Ne—Na2体系的二维等势图(图4),与图2的结果吻合,整个势能曲线在总体上显出相互作用弱,势能阱较浅,当Ne距离Na2分子比较远时,势能面以各向同性为主,仅当Ne靠近Na2分子时才出现弱的排斥现象.这种特性在大多数的多原子分子体系中常见[16-18].

图3 Ne—Na2体系在r=re时不同角度的能量对比
Figure 3Energy comparison of different angles whenr=rewith potential energy surface of Ne—Na2

图4 Ne—Na2(r=re)体系势能面等势面(等高线单位为cm-1)
Figure 4Contour plots of the potential for Ne—Na2system atr=re(Contours are labeled in cm-1).
图5是 0≤R≤20a0区域的全程三维势能曲线,由于相互作用较弱,势阱位置不明显.图6是0≤R≤10a0区域的势能曲线,这时图中势阱位置较清晰,整个势能面仍然呈现弱相互作用.在短程区域,主要由于Ne原子受到Na2双原子分子形成的电子云的排斥,所以呈现出很明显的排斥势;但是随着R的增大,Hartree-Fork排斥作用逐渐减弱,这时电子关联产生的色散吸引作用逐渐增强,吸引作用是主要作用.因此在Ne单原子与Na2双原子共同作用中,形成了短程排斥和长程区域的浅势阱特性.
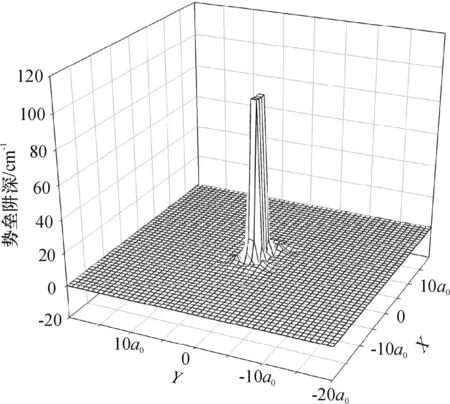
图5 Ne—Na2体系三维势能面远景
Figure 53D visions for the interaction potential of Ne—Na2system
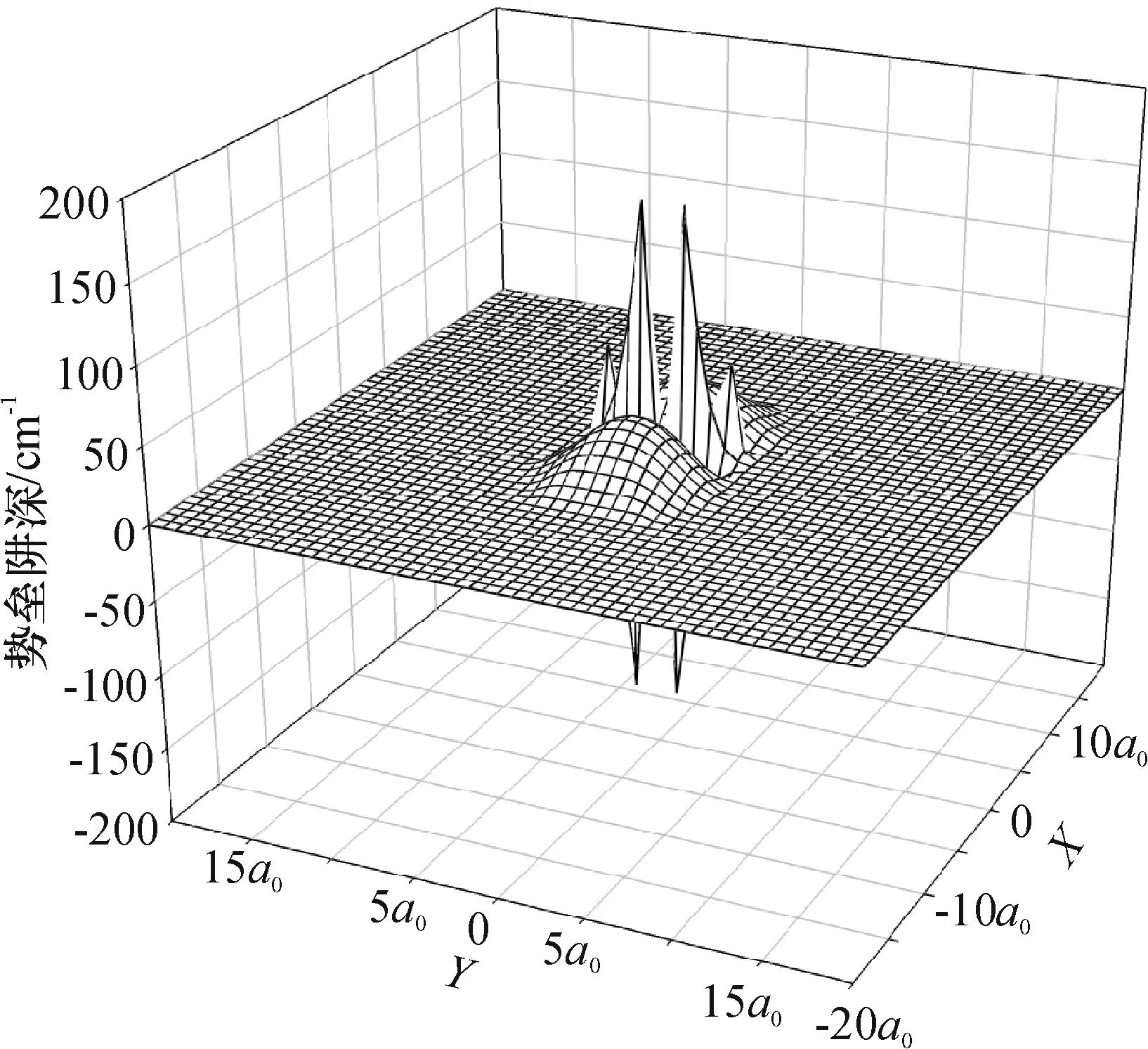
图6 Ne—Na2体系三维势能面近景Figure 6 3D close shot for the interaction potential of Ne—Na2 system
3结论
采用CCSD(T)方法及大基组,扫描了Ne—Na2体系的基态全程势能面;用最小二乘拟合方法绘制了其二维势能面和三维势能面,结果发现存在2个较浅的势阱:θ=80°,RNe—Na=3.3a0处,阱深约为-6.750×10-1cm-1和线型结构θ=0°,RNe—Na=10a0处,阱深约为-2.341×10-3cm-1.这些结果为进一步研究Ne—Na2体系的理论与实验的提供参考.
参考文献:
[1]邱英,何军,王彦华,等.三维光学晶格中铯原子的装载与冷却[J]. 物理学报,2008, 57(10):6227-6232.
QIU Y, HE J,WANG Y H,et al. Loading and colling of cesium atoms in 3D optical lattice[J].Acta Physica Sinica,2008,57(10):6227-6232.
[2]武宏宇,尹澜.超流费米气体相滑移时的密度分布[J].物理学报, 2006,55(2): 490-493.
WU H Y,YIN L.Density profile of a superfluid Fermi gas with a phase slip[J].Acta Physica Sinica,2006,55(2):490-493.
[3]WEINSTEIN J D,DE CARVALHO R,GUILLET T,et al.Magnetic trapping of calcium monhydride molecules at millikelvin temperatures[J].Nature,1998,395(10):148-150.
[4]BETHLEM H L,BERDEN G,MEIJER G.Decelerating neutral dipolar molecules[J].Physical Review Letters,1999,83(8):1558-1561.
[5]FIORETTI A,COMPARAT D,CRUBELLIER A.Formation of cold Cs2molecules through photo association[J].Physical Review Letters,1998,80(20):4402-4405.
[6]DONLEY E A,CLAUSSEN N R,THOMPSON S T.Atom-molecule coherence in a Bose-Einstein condensate[J].Nature,2002,417(6888):529-533.
[7]JOCHIM S,BARTENSEIN M,ALTMEYER A.Bose-Einstein condensation of molecules[J].Science,2003,302(19):2101-2103.
[8]GREINER M,REGAL C A,JIN D S.Emergence of a molecular Bose-Einstein condensate from a Fermi gas[J].Nature,2003,426(26):537-540.
[9]HEFTER U,JONES P L,MATTHEUS A,et al.Resolution of supernumerary rotational rainbows in Na2Ne Scattering[J].Physical Review Letters,1981,46(14):915-918.
[10]臧华平,李文峰,令狐荣锋,等.20Ne(34Ne)原子与18Na2(23Na2,37Na2)分子低温下冷碰撞的同位素效应研究[J].物理学报,2011,60(5):303-311.
ZANG H P, LI W F, LINGHU R F, et al. Study of isotope effect in20Ne(34Ne)-18Na2(23Na2,37Na2) low-temperature collisions[J]. Acta Physica Sinica, 2011, 60(5): 303-311.
[11]BERGMANN K,ENGELHARDT R,HEFTER U,et al.State-resolved differential cross sections for rotational transitions in Na2+Ne(He) collisions[J].Physical Review Letters,1978,40(22):1446-1450.
[12]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 09,Revision D.01[EB/OL].(2015-03-18)[2015-03-29]. http://www.gaussian.com/citation_g09.htm.
[13]THOM H. DUNNING J.The Cl+Bk extrapolation method. Application to hydrogen fluoride[J].Chemical Physics,1970,42(3):251.
[14]BOYS S F, BERNARDI F.Calculation of small molecular interactions by differences of separate total energies some procedures with reduced errors[J].Molecular Physics,1970,19(4):559.
[15]GAO A F, LIANG X L,LI L H,et al.A Gaussian-3 theoretical study of the alkylthio radicals and their anions: structures,thermo chemistry,and electron affinities[J].Journal of Molecular Modeling,2013,19(8):3226.
[16]杨洋.NH2+H2→NH3+H反应的从头算势能面及量子动力学研究[D].武汉:中国科学院武汉物理与数学研究所,2013.
[17]WANG Y H,XIAO C Y,DENG K M,et al.Quasi-classical trajectory study of the isotope effect on the stereodynamics in the reaction H(~2S)+CH(X~2Π;u=0,j=1)→C(~1D)+H2(X~1Σg~+)[J].Chinese Physics B,2014,32(4):240-245.
[18]YUE X F.Product polarization and mechanism of Li+HF(v=0,j=0)→LiF(v′,j′)+H collision reaction[J].Chinese Physics B,2013,22(11):275-281.
【中文责编:谭春林 英文责编:肖菁】
AbinitioCalculation and Analytic Fits on the Potential Energy Surface for Ne—Na2Complex
WANG Yue1*, LI Weiyan1, WANG Xiaorun1, ZHANG Fei1, ZHANG Qiang1, FANG Fang2
(1.Department of Electrical Engineering, Tongling University, Tongling 244000, China;2.Tongling Environmental Protection Agency, Tongling 244000, China)
Abstract:By using the aug-cc-pVQZ(Augmented versions of the preceding basis sets with added diffuse functions) /Na, aug-cc-pVDZ(Augmented versions of the preceding basis sets with added diffuse functions)/Ne and the bond functions (bf) basis sets, three-dimensional potential energy surfaces (PES) for the interaction of rigid Na2with Ne are calculated.The coupled cluster singles-and-doubles with noniterative inclusion of connected triple [CCSD (T)] level of theory are used.The 146 ab initio points on the PES are fitted to a 96-parameter. The results of the interaction energy calculations and the corresponding fits to analytical functions are presented.Contour plots also are given. The deeper potential well is θ=80°,RNe—Na=3.3a0 with well depth -6.750×10-1cm-1. The linear configuration with well depth -2.341×10-3cm-1is at θ=0°,RNe—Na=10a0. The whole potential energy surface exhibits weak anisotropy.
Key words:Ne—Na2; CCSD(T); ground state of molecules; potential energy surface; ab initio calculation
中图分类号:O561.1
文献标志码:A
文章编号:1000-5463(2016)01-0035-05
*通讯作者:王悦,副教授,Email:wangyue8001@qq.com.
基金项目:国家自然科学基金(11205115);安徽省高等学校省级自然科学研究项目(KJ2013B303);2015年安徽省大学生创新性训练计划项目(201510383075,201510383073,201510383074,201510383132)
收稿日期:2015-03-29《华南师范大学学报(自然科学版)》网址:http://journal.scnu.edu.cn/n
