G蛋白偶联受体计算研究的进展和前瞻
2016-04-28许伟明王晓锋蔡伟文鄢仁祥
许伟明,王晓锋,林 娟,3,蔡伟文,鄢仁祥,3*
(1.福州大学生物科学与工程学院,福州 350108;
2.山西师范大学数学与计算机科学学院,山西 临汾 041004;
3.福建省海洋酶工程重点实验室,福州 350108)
G蛋白偶联受体计算研究的进展和前瞻
许伟明1,王晓锋2,林娟1,3,蔡伟文1,鄢仁祥1,3*
(1.福州大学生物科学与工程学院,福州 350108;
2.山西师范大学数学与计算机科学学院,山西 临汾 041004;
3.福建省海洋酶工程重点实验室,福州 350108)
摘要:G蛋白偶联受体(G protein-coupled receptor, GPCR)是含有七个跨膜螺旋的一类重要蛋白,是迄今为止发现的最大的多药物靶标受体超蛋白家族。例如,目前上市药物中有超过30%是以GPCR为靶点的。然而,与GPCR重要性形成强烈反差的是科学界对于其结构与功能的了解非常贫乏,主要原因是通过实验手段来获得GPCR的结构与功能信息极其困难。利用生物信息学方法从基因组规模的数据中识别GPCR并预测三维结构是可行途径之一。基于生物信息学的GPCR研究将为新型药物靶标的筛选和药物的开发提供一定的帮助。本文论述了几种较为典型的GPCR计算方法,并基于已有研究提出可能的创新性研究策略来解决GPCR蛋白识别、跨膜区定位、以及结构和功能预测等问题。
关键词:G蛋白偶联受体;GPCR识别;蛋白结构预测;跨膜区预测;药物配体
1前言
生物学中的受体(Receptor)一般是指一类介导细胞信号转导的功能蛋白,能识别并结合周遭环境中的某些微量物质后通过信号放大系统触发后续一系列生理和生化反应[1, 2]。G蛋白偶联受体是一大类膜蛋白受体的统称,它们可以把各种各样的胞外信号传递到细胞内,并通过和其它信号转导通路间的相互作用调节各种生物学功能。GPCR同时是重要的药物靶标蛋白,广泛地应用在各种医学治疗领域,比如抑郁症、疼痛、肥胖症、哮喘、焦虑症、高血压、癌症、心血管疾病、帕金森症、糖尿病,精神分裂症等。至今已知的GPCR药物中有739种可用于治疗疼痛、486种治疗哮喘、480种治疗高血压,上市的药物有648种,而处于各种研发阶段的GPCR药物超过6 600种[3]。GPCR药物按其作用机理可分为激动剂、拮抗剂、反向激动剂、调节剂等受体[4, 5]。因其在药物研发和科学研究方面的巨大潜在价值而受到科学界的极大关注。GPCR的立体结构一般由膜外N末端、7个跨膜α螺旋(Trans Membrane Helix,TM1-TM7)、3个胞外环(Extra Cellular Loop,ECL1-ECL3)、3个胞内环(Intra Cellular Loop,ICL1-ICL3)以及膜内C末端组成。被七条跨膜螺旋反复穿过的细胞膜的脂双层,肽链的C端以及ICL3上都有G蛋白的结合位点[6, 7]。图1是一个PDB编号为4UHR的GPCR蛋白结构,该结构中有7个明显的跨膜α螺旋。
GPCR同时是人体内膜蛋白家族中成员数最多的一个种类,在人体基因组编码的蛋白中约有800~1000个GPCR[8],包括A、B、C、E、F五大类,其中D类只存在于酵母等少数低等真核生物体中。GPCR在信号传递中发挥着重要作用,普遍具有激活G蛋白的能力,可介导多种生物学功能。在静息状态下,
GPCR在膜上与由Gα、Gβ和Gγ三个亚基组成的异三聚体G蛋白结合形成复合物。其中Gα亚基上结合有GDP分子。当GPCR与胞外配体结合后发生构象变化(GPCR被激活),活化的受体会催化Gα亚基捕获GTP 分子来交换先前结合的GDP,GTP与Gα亚基的结合会使受体与G蛋白的复合物解离,受体、GTP-Gα和Gβ-Gγ二聚体三者相互分开。GTP-Gα激活腺苷酸环化酶,酶C或离子通道等,继而激活下游的信号通路,包括甘油二酯(Diacyl glycerol,DG)、三磷酸肌醇(Inositol trisphosphate,IP3)、钙信号和第二信使cAMP等(如图2所示)。

图1 4UHR蛋白三级结构Fig.1 Tertiary structure of protein 4UHR

图2 GPCR介导的一种细胞信号传导机制示意图Fig. 2 Sketch of GPCR-mediated cell signaling mechanisms
后两者可以进一步与其它蛋白相互作用从而使信号继续传递下去,而自由的受体可以重新结合上一个新的G蛋白来开始下一轮信号转导过程。现在已知的GPCR配体有光、气味、激素、趋化因子与神经递质等[9]。这些配体可以是某些小分子物质如糖类、多肽和脂质,也可以是蛋白质等生物大分子。部分GPCR的配体结合部位处于跨膜螺旋和胞外环附近,不过也有一些例外,如富亮氨酸重复G蛋白偶联受体和糖蛋白激素受体等。其它类型的GPCR则主要以N端与配体结合。 也有一些报道指出B类受体的跨膜螺旋上也存在潜在的变构配体结合位点。近年来提出的GPCR功能选择理论预示不同配体可以诱导不同GPCR 构象,从而选择性地激活下游的信号转导通路。美国科学家RobertJ.Lefkowitz与BrianK.Kobilka,因为突破性地揭示了GPCR的内在工作机制,而获得了2012年诺贝尔化学奖。GPCR的研究成为不少国际药物公司竞争的重点。据估计,目前国际医学市场上可以获得的药物中,有30%以上临床用药是与GPCR作为药物靶标直接相关的。全球最畅销的20种药物中,以GPCR为作用靶标的达到60%,每年的销售总额高达500亿美元[3]。但是,与其重要性形成强烈反差的是现今科学界对于GPCR的结构与功能了解极其贫乏。例如,截止到2015年8月,蛋白质数据库PDB[10]中存储的GPCR晶体结构仅有125个,结构数量的缺乏严重地制约了基于结构信息对GPCR进行功能、小分子结合物与配体的研究。另外,虽然在GPCRDB(http://www.gpcr.org/7tm/)[11]数据库中存有30 569 条GPCR序列,但是其中至少70%以上为功能未知的序列,同时大多数GPCR 参与的都是网络状的复杂生理和生化过程,这给以GPCR为靶标的药物研发带来了巨大挑战。GPCR结构、功能及代谢过程数据的缺乏有其客观原因:(1)天然GPCR含量低导致难以用基因工程方法大量表达;(2)GPCR 不易在活性形态下分离、纯化;(3)膜环境的特性使其结晶有很大困难;(4)实验研究GPCR需要投入大量人力和科研经费。表1列出了部分GPCR相关的生物信息学工具,虽然在GPCR生物信息研究方面欧美等发达国家一直处于领先地位,但令人欣慰的是近年来我国也正在加快步伐开展研究。例如:2014年,中国科学院上海药物研究所赵强研究组与美国Scripps研究所,美国国立卫生研究院(NIH) 和德国波恩大学通力合作,首次解析了P2Y12R受体与抗血栓药物AZD1283复合物的高分辨率的晶体结构,并于2014年5月1日在 Nature 杂志发表题为“Structure of the human P2Y12 receptor in complex with an antithrombotic drug”的研究文章[2]。中国科学院生物物理研究所蒋太交研究组也在蛋白质结构预测及膜蛋白的研究方面取得了一定的研究成果。另外,清华大学、北京大学和中国科学院上海生命科学研究院等高等学府和研究机构也都在膜蛋白生物信息学研究领域投入了越来越多的科研资源。
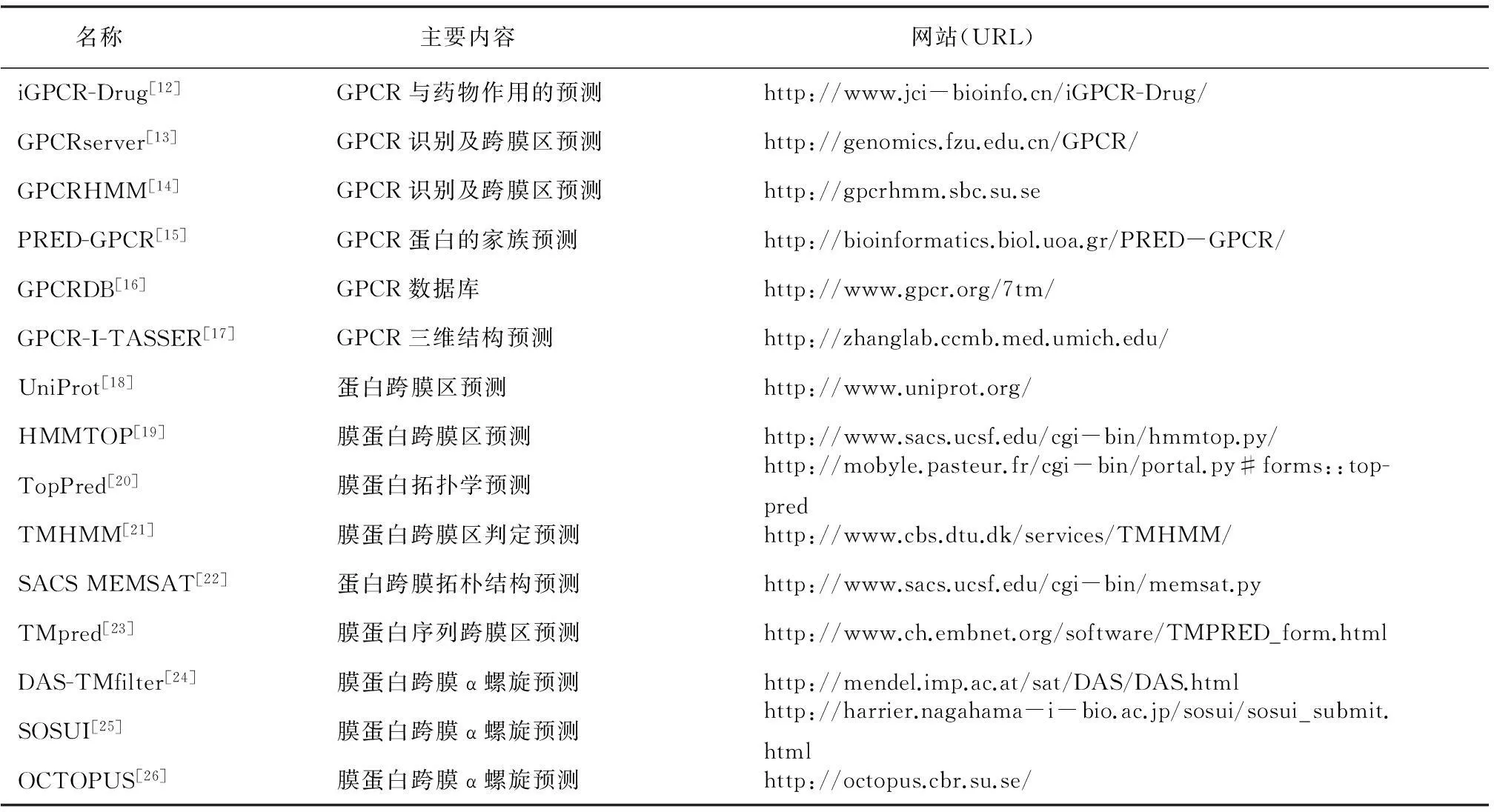
表1 国际GPCR相关的生物信息学程序/服务器/数据库
2GPCR的计算研究
蛋白质的一级序列决定三维结构信息,而其三维结构一定程度上决定其生物学功能。为更好了解GPCR的功能,测出其三维结构是重要的途径之一。由于GPCR是七跨膜螺旋的膜蛋白,很难得到晶体,通过X射线衍射确定其三维空间结构, 同样也很难在水溶液中用核磁共振方法获得它的动态结构[27],因此通过实验解析GPCR三维结构极具挑战性。然而统计分析发现GPCR一般具有保守的跨膜螺旋结构,序列特征明显,比较适合用生物信息学预测的方法定位其跨膜螺旋区的位置。所以目前学术界的研究趋势是通过开发相应的生物信息学工具来尝试在基因组规模上研究GPCR。关于GPCR的现有生物信息学研究,主要集中在三个问题上:(1) GPCR蛋白的识别;(2)GPCR跨膜区的预测;(3) GPCR功能和药物配体结合位点预测。
2.1GPCR蛋白的识别
基于序列相似性识别GPCR的方法[28, 29],其出发点是基于功能序列的保守性,通过序列比对工具BLAST等,
从非冗余核酸序列、表达序列标签(EST)、蛋白质序列里面挖掘出可能的GPCR序列。当发现一个新的序列与已存在的GPCR序列有足够的相似度时,再通过对跨膜区的分析来识别GPCR,或发现新的GPCR亚家族。该方法的不足是海量的待预测序列导致计算量比较大,结果分析较为繁琐,得到的结果准确度不高。另一种替代方法是通过预测软件找出所有可能为GPCR序列的开放阅读框(ORF),将已知蛋白序列的GPCR排除出去,再将剩余的未知ORF序列作一个数据库,用已知GPCR序列集对数据库进行BLAST比对分析。但是此方法也存在一定局限性:因为各种预测软件的精度不高,此方法对它们有强依赖性,其预测结果会直接影响到跨膜螺旋区的预测分析。
基于序列信息的统计特征识别GPCR,其中比较有代表性的是SAM-T2K和T-HMM方法。 Karchin等[30]通过SAM-T2K算法对属于同一个GPCR家族的序列进行多序列比对,再通过隐马尔可夫模型(Hidden Markov Model,HMM)的方法构建能代表这个家族的统计模型,最后分别计算这些模型的概率并将其转化为固定长度的特征向量( Fisher Score Vector,FSV),构建支持向量机(SVM)家族分类器,对GPCR进行分类识别。此方法中HMM模型的长度是很重要的参数,这个参数表示主状态的数目,它会直接影响分类的结果。不同的方法得到的预测结果可能都不相同。Qian等[31]使用T-HMM方法,对GPCRs构建系统进化树,然后对系统进化树上每一个节点和子节点通过HMM算法对不同家族和亚家族进行建模,依据T-HMM的最高分值来判定未知序列而识别GPCR。这个方法存在的问题是:T-HMM方法应用的前提是认定同一类型的配体其结合的GPCRs序列应该在进化距离上是相近的。但是从配体结合的角度,与同一类配体相结合的序列会有较大差异,相应的进化信息就体现的不够明显,因此该方法只能适用于进化距离较近的情况。对于T-HMM方法的不足,Bhasin等[32]利用双联氨基酸的使用频率结合SVM以及氨基酸的部分物理化学性质如电荷、极性、范德华力、疏水性等特性解决了对一些进化距离较远的GPCR蛋白的识别问题。
2.2GPCR跨膜区的预测
GPCR 蛋白嵌在生物膜中,这使得GPCR蛋白有着与球蛋白不同的生物化学特性。因此,准确地获得GPCR蛋白扩膜区与非跨膜区的信息对判断GPCR的生物学功能起到关键的辅助作用[33]。GPCR跨过磷脂双分子层,这意味着其跨膜区都是由强的疏水性氨基酸组成,而磷脂双分子层的厚度决定了每次跨膜的氨基酸大约为20个左右[34]。20世纪80年代初kyte,Doolittle等[35]提出了氨基酸疏水标度值,根据这个疏水标度值,将氨基酸序列依次通过一个长方形的框架,从而转换成疏水图谱,设定恰当的阈值,从而寻找跨膜区。80年代中期Von Heijine等[20]发现“正电荷居内规则”即对所有已知跨膜蛋白进行统计分析,发现跨膜区的内膜周围氨基酸都是带正电荷的。90年代初,首次将“正电荷居内规则”和疏水性分析结合,开发跨膜区预测工具TopPred大大提高了跨膜区预测精度。90年代末,开发的MEMSAT预测工具将跨膜蛋白的氨基酸按照在跨膜核心区、膜内外出现的频率,以及在跨膜区末端出现的频率与在整个跨膜蛋白出现频率之比,计算出氨基酸的偏好性。再将氨基酸的偏好性与动态规划算法结合起来预测蛋白质跨膜区。基于类似原理设计的TMHMM预测工具用HMM统计分析已知跨膜蛋白的跨膜区两端,跨膜核心区,膜内环、膜外环和长环以及远离膜区的氨基酸分布,算出每个氨基酸残基位于跨膜区、膜内外的概率进行跨膜蛋白的跨膜区预测。同样基于HMM的预测工具HMMTOP则通过统计分析五个不同状态组成的模型,即跨膜蛋白的跨膜核心区,膜内、外环,膜内螺旋及膜外螺旋的尾部氨基酸残基分布,同时基于蛋白质拓扑结构改变会直接导致氨基酸分布改变的基础上将氨基酸分布差异最大的组合状态考虑在内以预测跨膜区。随着人工神经元网络算法 (Artificial Neural Networks ,ANN)的发展,viklund等[26]通过对已知结构的跨膜蛋白统计分析氨基酸残基倾向性分数,再与HMM结合开发出OCTOPUS预测工具。近年随着ANN,HMM,支持向量机(SVM)[36]等的共同发展,跨膜蛋白跨膜区的预测精度一步步提高。表二,列出了目前可用于GPCR跨膜区预测的几种生物信息学工具,并选择PDB编号4UHR的GPCR蛋白测试。几种预测软件精确度有差异。现在多数科研工作者使用HMMTOP以及 TMHMM 2个开发工具,两者都是采用跨膜区为α螺旋的膜蛋白来训练模型的,而这些膜蛋白并非全是 GPCR,有些膜蛋白只有1个或者2个跨膜片段,这样导致在预测GPCR的跨膜区的性能可能达不到理论上的最优(见表2)。

表2 4UHR蛋白跨膜区预测结果
同时,GPCR-I-TASSER在预测GPCR的三维结构上,其优势在于其模拟过程中的片段组装算法,而在前期其使用的蛋白质折叠识别(Fold Recognition)[37]算法中并没有专门针对GPCR 这种类型的蛋白序列具有其特异性的比对算法。
2.3GPCR功能和药物配体结合位点预测
蛋白质的功能往往决定于其结构,在结构预测系统的基础上,利用结构信息预测GPCR的生物学功能,主要包括结合位点(Binding site)和结合底物(Ligand)的预测上。这部分将主要使用结构信息以及一些物理统计学的方法进行研究。
19世纪末统计与遗传学家Fisher[38]提出受体学说:受体与配体的识别关系犹如锁与钥匙的关系。随后20世纪中叶Koshland[39]在受体学说的基础上提出著名的诱导契合理论,表明配体分子并不是事先就以与受体互补的形态存在着,而是在受到诱导之后不断重置蛋白质受体的活性口袋,使两者结合的更为紧密。根据对空间和能量的不同处理方式,分子对接被分为以下三类[40, 41]:(1)刚性对接即在受体配体分子构像都不变化的前提下只变换对接分子的姿态和方位进行对接;(2)半柔性对接即固定受体分子的构像,变换配体小分子的构像而进行对接;(3)柔性对接即受体配体分子都能进行分子构像的变化而进行对接。GPCR蛋白与药物配体的分子对接,即在GPCR三维结构已知的情况下,在其活性部位依据空间、形状、性质互补的原则置入药物配体分子,形成具有特定关系的受体-配体复合物。基于受体结构的虚拟数据库筛选方法利用分子对接技术自动地匹配受体结合腔穴和化合物数据库中的小分子三维结构,然后利用基于分子力场的能量函数或者经验性函数对分子对接的模式进行打分,进而选择与受体相互作用最好的一组化合物进行生物活性测试,从而大大节省了寻找先导化合物的费用和难度。尽管分子对接在先导化合物的寻找方面有许多成功的应用,但仍然存在很大的问题,其中忽略蛋白柔性常常是导致失败的重要原因。为此急需建立一个专门用于研究配体结合位点柔性的关系型数据库,可助于研究蛋白质配体结合位点的构象变化和蛋白质与配体之间的相互作用,以及两者之间的关系;同时提供配体结合位点的多个不同三维构象、结合位点残基的物理和化学的性质以及蛋白质和配体相互作用的描述符等。
3GPCR预测算法改进的可能策略
3.1优化的GPCR识别打分函数
一种改进的GPCR识别方法:通过深入分析GPCR弱同源蛋白序列和结构的进化关系的基础上,开发合适的打分函数(能量函数),用来评判两个GPCR蛋白之间的弱同源性。然后依据该打分函数,对未知序列通过搜索构建好的GPCR数据库,寻找合适的结构(弱)同源GPCR蛋白,之后预测查询蛋白是否为GPCR。构建的GPCR识别能量函数是采用动态打分的方式。如果待预测的序列与GPCR模板数据库中的模板存在较高的序列相似度,则采用更多的序列方面的信息进行打分,这样可以避免相应结构性质预测不准确时带来的噪音;如果待预测的序列与GPCR模板数据库中的模板存在相似度非常低,则可以计算出相似度数值,同时预测出待预测的序列的结构性质(包括氨基酸深度、表面溶剂可及性以及二面角等信息),可以更依赖这些结构性质进行打分。GPCR膜蛋白存在结构核心区和结构可变区。通过结构比对软件,寻找不同GPCR家族之间的保守区域,把这部分的数据作为开发GPCR生物信息学工具的辅助数据库。通过文献挖掘,把文献中报道的相对稳定区域与重要功能区域及位点等信息加入到该辅助数据库中。在开发新算法时充分突出结构核心区的重要性,这样处理将较大程度地提高GPCR识别能量函数的性噪比,同时提高预测性能。
3.2基于分割片段的GPCR跨膜区预测算法
对于GPCR跨膜区预测工具的发展和存在的不足,我们建议开发专门的GPCR跨膜区预测算法,构建一个跨膜区片段的辅助数据库,通过Profile-Profile比对算法,把这些跨膜区片段比对到未知序列中去。根据片段比对的结果比较准确地去预测未知序列的跨膜区。同时构建一些具有互补性的编码。从序列谱(Profile)提取出20种氨基酸出现的位置信息(Position-specific scoring matrix)及k-空格氨基酸对信息(K-spaced residue pair composition)。序列谱的位置信息和 k-空格氨基酸对组成信息具有非常好的互补性,再结合人工神经元网络算法将提高GPCR跨膜区预测的准确率。
增强的 Profile-Profile 比对算法,可以用于改进GPCR弱同源序列间的比对精度。GPCR在跨膜区相对保守,采用多结构比对软件来寻找GPCR的其它潜在保守区域。在这些保守的区域中加大比对算法中的空位罚分(Gap penalty),同时在打分函数中考虑跨膜区的影响。在获得初步的比对结果后依据氨基酸的理化性质和PSI-Blast搜索得到的多序列比对结果中对Profile-Profile的比对结果进行深度优化,可使比对的结果更加准确,虽然可能会消耗更多的计算时间及资源。但当前可获得的GPCR晶体结构数据比较少,这过程中多消耗的计算时间及资源是在可接受的范围内,Profile-Profile 的比对打分函数 S(i,j)为:
S(i,j)=Profile(i,j)+w1SS_Sim(i,j)+
(1)
其中 Profile(i,j)为常规的序列谱与序列谱相似性函数,采用点积(Do-product)或者皮尔逊相关函数;SS_Sim(i,j)为二级结构元素的相似性,简单地采取相同的二级结构打分记为+1;不相同的二级结构记为-1;为两个蛋白不同结构特性之差,同时可以采用一些新的结构性质提高比对准确性;shift参数用于调整比对的总体分数,以防止不相似的片段比对上。与其他方法相比,用分割好的模板的跨膜区片段对未知查询序列进行基于片段的Profile-Profile比对,可以很好地识别未知查询序列的跨膜区。另外,对已有清晰跨膜区的GPCR进行片段分割,建立GPCR跨膜区片段数据库,同时根据针对该片段数据库训练好的Profile-Profile比对算法,来准确地把跨膜片段比对到未知序列的相应位置上。若未知序列为GPCR,则比对的结果本身很可能就有七个潜在的跨膜区。目前基于片段比对的跨膜区预测算法目前在学术界还较少有报道。
3.3综合应用具有互补性的特征编码
开发一系列具有互补性的编码(Encoding), 同时使用已构建好的人工神经元网络算法(ANN)对编码进行建模。可以从序列谱(Profile)提取出20种氨基酸出现的位置信息以及k-空格氨基酸对信息,这些编码的详细计算过程可查询文献[42, 43]。这些具有20种氨基酸的位置信息与组成信息的编码具有非常好的互补性,在外膜蛋白的识别中与GPCR预测中这些编码都有效。利用人工神经元网络算法,其特色在于结合遗传算法与后向传播算法来优化权重,同时在不同的迭代过程中更新权重的学习率(即 Learning rate)采取动态的变化。以k-空格氨基酸对信息作为输入,人工神经元网络程序在取得较优化的权重模型时会取得比支持向量机相当或者更好的预测结果。
4结语与展望
GPCR作为最大的多药物靶标受体超家族,目前超过30%的上市药物以其为靶点,但通过实验途径获得的GPCR结构与功能数据却很少。令人欣慰的是,现在通过生物信息学的方法来研究GPCR的结构与功能特征已得到较好地开展。本文综述了GPCR的研究现状,同时讨论了GPCR识别和结构预测的生物信息学研究以及急待解决的一些潜在问题,提出一些创新性的可能策略。通过开发全新的GPCR识别打分函数、基于分割片段的GPCR跨膜区预测算法以及增强的Profile-Profile比对算法来改进GPCR识别、跨膜区预测以及与药物配体结合的预测精度,有助于解决实验科学家在进行GPCR研究过程中遇到的问题,增强对GPCR蛋白、结构和功能关系的认识,对功能基因组学,药物研发等实验研究提供强有力的技术和理论支持。相信在更多研究者的不懈努力下,揭开GPCR的神秘面纱指日可待,为GPCR新型药物靶标的筛选和药物的开发研究拓展广袤边疆。
参考文献
[1]NGLESE J, KOCH W J, CARON M G,et al. Isoprenylation in regulation of signal transduction by G-protein-coupled receptor kinases[J]. Nature, 1992,359(6391):147-150.
[2]ZHANG K, ZHANG J, GAO Z G,et al. Structure of the human P2Y12 receptor in complex with an antithrombotic drug[J]. Nature, 2014,509(7498):115-118.
[3]赵强, 吴镭, 李佳,等. 重大疾病导向的G蛋白偶联受体研究[J]. 中国基础科学, 2015,(03):3-8.
ZHAO Qiang,WU Lei, LI Jia,et al.Carbon budget of forest ecosystems and its driving forces[J].China Basic Science,2015,(03):3-8.
[4]KRATOCHWIL N A, GATTI-MCARTHUR S, HOENER M C,et al. G protein-coupled receptor transmembrane binding pockets and their applications in GPCR research and drug discovery: a survey[J]. Current Topics in Medicinal Chemistry, 2011,11(15):1902-1924.
[5]XIAO X, MIN J L, WANG P,et al. iCDI-PseFpt: identify the channel-drug interaction in cellular networking with PseAAC and molecular fingerprints[J]. Journal of Theoretical Biology, 2013,337:71-79.
[6]KIM S, MALINVERNI J C, SLIZ P,et al. Structure and function of an essential component of the outer membrane protein assembly machine[J]. Science, 2007,317(5840):961-964.
[7]PIERCE K L, PREMONT R T, LEFKOWITZ R J. Seven-transmembrane receptors[J]. Nature Reviews Molecular Cell Biology, 2002,3(9):639-650.
[8]VASSILATIS D K, HOHMANN J G, ZENG H,et al. The G protein-coupled receptor repertoires of human and mouse[J]. Proceedings of the National Academy of Sciences , 2003,100(8):4903-4908.
[9]李静, 谢欣.靶向G蛋白偶联受体的高通量药物筛选方法[J].国际药学研究杂志,2012,39:353-357.
LI Jing,XIE Xin.High-throughput screening assays for G-protein- coupled-receptors-targeted drug discovery[J].Journal of International Pharmaceutical Research,2012,39:353-357.
[10]BERMAN H M, BHAT T N, BOURNE P E,et al. The Protein Data Bank and the challenge of structural genomics[J]. Nature Structural Biology, 2000,7 Suppl:957-959.
[11]HORN F, BETTLER E, OLIVEIRA L,et al. GPCRDB information system for G protein-coupled receptors[J]. Nucleic Acids Research, 2003,31(1):294-297.
[12]XIAO X, MIN J L, WANG P,et al. iGPCR-drug: a web server for predicting interaction between GPCRs and drugs in cellular networking[J]. PLoS One, 2013,8(8):e72234.
[13]YAN R, WANG X, HUANG L,et al. GPCRserver: an accurate and novel G protein-coupled receptor predictor[J]. Molecular BioSystems, 2014,10(10):2495-2504.
[14]WISTRAND M, KALL L, SONNHAMMER E L. A general model of G protein-coupled receptor sequences and its application to detect remote homologs[J]. Protein Science, 2006,15(3):509-521.
[15]PAPASAIKAS P K, BAGOS P G, LITOU Z I,et al. PRED-GPCR: GPCR recognition and family classification server[J]. Nucleic Acids Research, 2004,32(Web Server issue):W380-W382.
[16]HORN F, BETTLER E, OLIVEIRA L,et al. GPCRDB information system for G protein-coupled receptors[J]. Nucleic Acids Research, 2003,31(1):294-297.
[17]ZHANG Y. I-TASSER: fully automated protein structure prediction in CASP8[J]. Proteins, 2009,77 Suppl 9:100-113.
[18]APWEILER R, BAIROCH A, WU C H,et al. UniProt: the Universal Protein knowledgebase[J]. Nucleic Acids Research, 2004,32(Database issue):115-119.
[19]TUSNADY G E, SIMON I. Principles governing amino acid composition of integral membrane proteins: application to topology prediction[J]. Journal of Molecular Biology, 1998,283(2):489-506.
[20]VON HEIJINE G. Membrane protein structure prediction hydrophobicity analysis and the positive-inside rule[J].Journal of Molecular Biology, 1992,225(2):487-494.
[21]KROGH A, LARSSON B, VON HEIJNE G,et al. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes[J]. Journal of Molecular Biology, 2001,305(3):567-580.
[22]JONES D T. Improving the accuracy of transmembrane protein topology prediction using evolutionary information[J]. Bioinformatics, 2007,23(5):538-544.
[23]HOFMANN K, W S. Tmbase-A database of membrane spanning proteins segments[J]. American Journal of Respiratory & Critical Care Medicine,1993,374:166-171.
[24]HOFMANN K, W S. Organelle-specific expression of subunit ND5 of human complex I (NADH dehydrogenase) alters cation homeostasis in Saccharomyces cerevisiae[J]. Fems Yeast Research,1997,10(6):673 - 676.
[25]HIROKAWA T, BOON-CHIENG S, MITAKU S. SOSUI: classification and secondary structure prediction system for membrane proteins[J]. Bioinformatics, 1998,14(4):378-379.
[26]VIKLUND H, ELOFSSON A. OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar[J]. Bioinformatics, 2008,24(15):1662-1668.
[27]吴宏杰, 吕强, 权丽君,等.GPCR跨膜螺旋的结构拓扑建模及其预测方法[Z].计算机学报,2013,36(10):2168-2178.
WU Hongjie,L(U) Qiang,QUAN Lijun,et al.Modeling the structural topology and predicting the three-dimensional structure for transmembrane helixes of GPCR[Z].Chinese Journal of Computers, 2013,36(10):2168-2178.
[28]TAKEDA S, KADOWAKI S, HAGA T,et al. Identification of G protein-coupled receptor genes from the human genome sequence[J]. Federation of European Biochemical Societies Letters, 2002,520 (1-3):97-101.
[29]FREDRIKSSON R, SCHIOTH H B. The repertoire of G-protein-coupled receptors in fully sequenced genomes[J]. Molecular Pharmacology, 2005,67(5):1414-1425.
[30]KARCHIN R, KARPLUS K, HAUSSLER D. Classifying G-protein coupled receptors with support vector machines[J]. Bioinformatics, 2002,18(1):147-159.
[31]QIAN B, SOYER O S, NEUBIG R R,et al. Depicting a protein's two faces: GPCR classification by phylogenetic tree-based HMMs[J]. Federation of European Biochemical Societies Letters, 2003,554(1-2):95-99.
[32]BHASIN M, RAGHAVA G P. GPCRpred: an SVM-based method for prediction of families and subfamilies of G-protein coupled receptors[J]. Nucleic Acids Research, 2004,32(Web Server issue):W383-W389.
[33]WISTRAND M, KALL L, SONNHAMMER E L. A general model of G protein-coupled receptor sequences and its application to detect remote homologs[J]. Protein Science, 2006,15(3):509-521.
[34]ROSE G D. Prediction of chain turns in globular proteins on a hydrophobic basis[J]. Nature, 1978,272(5654):586-590.
[35]KYTE J, DOOLITTLE R F. A simple method for displaying the hydropathic character of a protein[J]. Journal of Molecular Biology, 1982,157(1):105-132.
[36]KARCHIN R, KARPLUS K, HAUSSLER D. Classifying G-protein coupled receptors with support vector machines[J]. Bioinformatics, 2002,18(1):147-159.
[37]JONES D T, TAYLOR W R, THORNTON J M. A new approach to protein fold recognition[J]. Nature, 1992,358(6381):86-89.
[38]FISCHER E. Einfluss der configuration auf die Wirkung der enzyme[J]. Berichte der Deutschen Chemischen Gesellschaft, 2006,27 (3):2985-2993.
[39]KOSHLAND D J. Correlation of structure and function in enzyme action[J]. Science, 1963,142(3599):1533-1541.
[40]EHRLICH L P, NILGES M, WADE R C. The impact of protein flexibility on protein-protein docking[J]. Proteins, 2005,58(1):126-133.
[41]MEILER J, BAKER D. ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility[J]. Proteins, 2006,65 (3):538-548.
[42]WANG X B, WU L Y, WANG Y C,et al. Prediction of palmitoylation sites using the composition of k-spaced amino acid pairs[J].Protein Engineering Design & Selection,2009,22(11):707-712.
[43]CHEN Z, CHEN Y Z, WANG X F,et al. Prediction of ubiquitination sites by using the composition of k-spaced amino acid pairs[J].PLoS One, 2011,6(7):e22930.
Progresses and prospects of computational study on G protein-coupled receptors
XU Weiming1, Wang Xiaofeng2,Lin Juan1,3, CAI Weiwen1,YAN Renxiang1,3*
(1.CollegeofBiologicalSciencesandEngineering,FuzhouUniversity,InstituteofAppliedGenomics,Fuzhou350108,China;2.CollegeofMathematicsandComputerScience,ShanxiNormalUniversity,Linfen041004,China;3.FujianKeyLaboratoryofofMarineEnzymeEngineering,Fuzhou350108,China)
Abstract:G protein coupled receptors (GPCR), a general designation of a large class of membrane proteins, contain seven transmembrane helices in its three-dimensional structure, which currently are the drug targets more than 30% in the market. In contrast to the importance of GPCR, the knowledge of scientific community to understand its structure and function is very limited. The main reason is the difficulty to obtain the structure and function of GPCR information by wet experiment. Now, it is feasible to use bioinformatics methods to identify and predict the 3D structure of GPCR. Research on GPCR based on bioinformatics is beneficial to novel drug targets screening and new drugs developing. This paper discusses some typical bioinformatics methods. In addition, several possible new research strategies are presented to address the identification of GPCR proteins from a genome scale database, position its transmembrane region and predict the three-dimensional structure of GPCR and drug ligand binding mode.
Keywords:G protein-coupled receptors; GPCR recognition; Protein structure prediction; Transmembrane region prediction; Drug ligand
中图分类号:Q51
文献标志码:A
文章编号:1672-5565(2016)01-031-08
doi:10.3969/j.issn.1672-5565.2016.01.06
作者简介:许伟明,男,硕士研究生,研究方向:生物信息学;E-mail:n140827017@fzu.edu.cn.*通信作者:鄢仁祥,硕士生导师,博士,研究方向:生物信息学;E-mail:yanrenxiang@fzu.edu.cn.
收稿日期:2015-12-29;修回日期:2016-3-1.
资助项目:国家自然科学基金青年项目(N0.31500673);福建省教育厅科技项目(N0.JA14049);福州大学人才基金项目(N0. XRC-1336)。
