基因序列的搜索与相似性比对
2016-04-25茹家康
茹家康,袁 琳
(1.辽宁大学 生命科学院,辽宁 沈阳 110036; 2.河南省人民医院 药学部, 河南 郑州 450003)
基因序列的搜索与相似性比对
茹家康1,袁琳2
(1.辽宁大学 生命科学院,辽宁 沈阳 110036; 2.河南省人民医院 药学部, 河南 郑州 450003)
摘要:生物信息学的基本任务是对各种生物分析序列进行分析,也就是研究新的计算机方法,从大量的序列信息中获取基因结构、功能和进化等知识,并将其存储于基因数据库中.而在序列分析中,将未知序列同基因数据库中已知序列进行相似性比较是一种强有力的研究手段,包括序列的片段测定、拼接,基因的表达分析,以及RNA和蛋白质的结构功能预测.
关键词:序列比对;分析;预测
1序列比对的相关概念
1.1序列的相似性与同源性
相似性是指一种很直接的数量关系,比如部分相同或相似的百分比或其他一些合适的度量.比如说,A序列和B序列的相似性是80%,这是个量化的关系.而同源性指从一些数据中推断出的两个基因或蛋白质序列具有共同祖先的结论,属于质的判断.就是说A和B的关系上,只有是同源序列,或者非同源序列两种关系.
序列的相似性和序列的同源性有一定的关系,一般来说序列间的相似性越高的话,它们是同源序列的可能性就更高,所以经常可以通过序列的相似性来推测序列是否同源.但实际上,只有序列是从一个不同祖先进化分歧而来,它们才是同源的;说序列共有50%的同源是没有意义的,而正确的应该是说它们有50%的相似度,并且可能是同源.
1.2序列比对
序列比对就是运用某种特定的数学模型或算法,找出两个或多个序列之间的最大匹配碱基或残基数,比对的结果反映了序列之间的相似程度以及它们的生物学特征[1].
1.2.1序列相似性
来自一个共同祖先的序列倾向于在序列、结构和功能上具有一定相似性,通过序列比对可以发现序列间的相似性,从而可以预测生物大分子的结构和功能.
1.2.2序列间比对的对应关系
在序列比对中可出现4种序列间比对的对应关系:Match (a,a): 匹配;Replace (a,b):替代;Delete (a,-): 缺失;Insert (-,b): 插入.
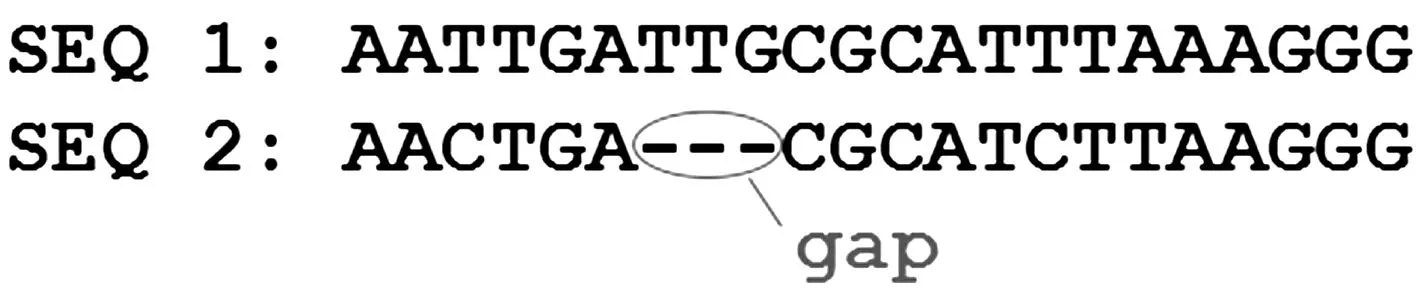
图1 gap举例Fig.1 Example of gap
按照进化的原则,对齐一致的碱基代表它们是祖先序列的一部分且仍然保持没有改变,对于没有对齐的序列,代表它们是有突变产生的,但是在不知道祖先序列的情况下,并不能确定突变发生在哪个序列上,祖先序列产生插入或缺失突变都可以产生gap(图1).
1.2.3序列比对模型分类
全局比对:从全局序列出发,考虑序列的整体相似性;局部比对:考虑序列部分区域的相似性.局部比对往往比全局比对具有更高的灵敏度,其结果更具生物学意义.
两个序列的联配叫做双序列比对,超过两个序列的联配叫做多序列比对,多序列比对可以更好地展示序列的保守性.

图2 打分矩阵Fig.2 Scoring Matrices
基因序列比对的方法有点阵法、动态规划算法,以及FASTA和BLAST等用于大数据的搜索程序.
1.2.4打分矩阵
要对两个序列进行排比,必须首先打出其相似性的定量分值,于是需要一个打分矩阵(图2).
打分矩阵的两个性质:①一致性(identity):两个蛋白质有一定数量的氨基酸在联配的位点上是相同的,即如果38个氨基酸的蛋白质中15个位点相同,其identity为 39.4%;②相似性(similarity):通常在某些位点上有一些氨基酸被另外一些化学物理特性相近的氨基酸所代替,这种突变可称为保守突变.将保守突变的因素考虑在内,就可以定义各种打分方案(scoring schemes)对两序列的相似程度打分,所得分值即代表其相似的程度.
1.2.5序列比对的统计检验
序列比对数学模型一般用来描述序列中每一个子字符串之间的匹配情况[2].通过改变某些参数可以得到不同比对结果,例如空位罚分值大小.此外,序列长度差异和字母表复杂度也会影响比对结果.合理调节参数,会减少空位数目,得到较好的结果,而放宽对空位罚分的限制,理论上任意序列都可以得到某个对比结果.因此序列比对的结果并不能作为两者之间一定存在同源关系的依据.
通常用序列比对程序给出一些统计值,用来表示结果的可信度.
2基因数据库
2.1核酸序列数据库GenBank
GenBank数据库包含了所有已知的核酸序列,以及与它们相关的文献著作和生物学注释.它是由美国国立生物技术信息中心(National Center for Biotechnology Information)建立和维护的(图3).GenBank数据以指数形式增长,核酸碱基数目大概每14个月就翻一倍.用户可以通过NCBI的主页使用GenBank.GenBank的宗旨是鼓励科研团体对DNA(Deoxyribonucleic Acid)序列的获取,从而促进数据库中DNA序列的丰富和更新,所以NCBI对GenBank的数据使用与发送没有任何限制.用户可从GenBank主页上下载Banklt(NCBI提供的WWW格式,用于便捷地提交DNA序列的数据)、Sequin(NCBI独立于操作系统的提交软件,可用于MAC、PC和UNIX平台,也可以通过FTP(File Transfer Protocol)远程获取)以及VecScreen(带菌污染物的筛选工具)等便于提交和更新研究成果的应用软件.
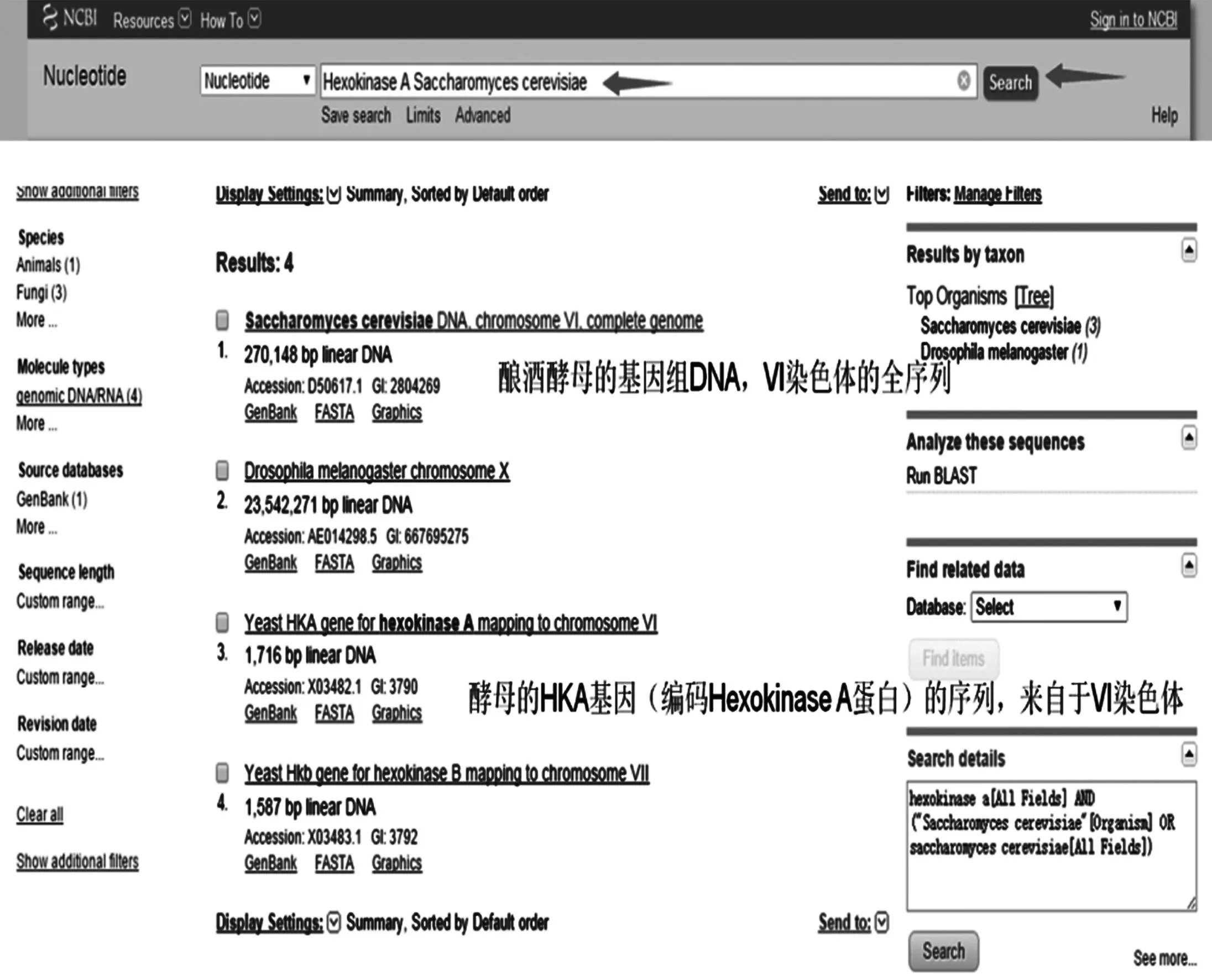
图3 GenBank界面Fig.3 Interface of GenBank
2.2蛋白质数据库
同一蛋白家族的多序列联配可以用来推断结构、功能和家族关键氨基酸的信息.多序列联配信息的表示方法有很多种,包括联配本身、一致序列、保守序列和残基模式、序列轮廓以及其他的序列家族的概率模型.
2.2.1PROSITE数据库
PROSITE是与蛋白质家族成员关系有关的序列模式数据库(图4).序列模式
[LIVM]-[ST]-A-[STAG]-H-C
代表有6个氨基酸残基组成的序列,[LIVM]表示L、I、V、M中的一个残基,[ST]表示S或T,接着是A,然后是S、T、A或G,再后是H,最后是C.
图4 PROSITE界面Fig.4 Interface of PROSITE
PROSITE模式也有很多缺点.首先,它们长度较短,不相关序列中有假阳性出现.其次,它们允许描述特定位置的变化,但无法计算该变化的概率.例如[LIVM]指某位点可能是L、I、V或M,但它没有说明L在家族90%的序列中出现,I、V、M仅出现在10%的序列中.
2.2.2PRINTS和BLOCKS
PRINTS和BLOCKS分别通过来自一组蛋白或蛋白家族中最高度保守区域的多序列联配无空位片段的形式表示蛋白质家族.这种多序列联配无空位片段分别定义为blocks(在BLOCKS中)或motifs(在PRINTS中).
例如SH3家族(蛋白质结构域)在PRINTS中用4个motif 表示,每个motif表示一个保守区域,因此PRINTS模式可以覆盖更大的序列区域,克服了PROSITE的部分缺点.与PROSITE不同,序列中motif的匹配通常要考虑氨基酸替换矩阵,不要求严格匹配.
3序列比对的应用
3.1BLAST程序
BLAST程序[3]是由NCBI开发的一个基于序列相似性的数据库搜索程序,其中包含了很多个独立的程序,这些程序是根据查询的对象和数据库的不同来定义的.比如说查询的序列为核酸,查询数据库亦为核酸序列数据库,那么就应该选择BLASTN子程序.
BLAST 具有非常广泛的应用, 查询序列可能具有某种功能,查询序列可能是来源于某个物种,查询序列可能是某种功能基因的同源基因,确定特定的蛋白质或核酸序列有哪些已知的直系同源或旁系同源序列.
BLAST程序评价序列相似性的两个数据:Score使用打分矩阵对匹配的片段进行打分,这是对各对氨基酸残基(或碱基)打分求和的结果,一般来说,匹配片段越长、 相似性越高则Score值越大.Evalue在相同长度的情况下,两个氨基酸残基(或碱基)随机排列的序列进行打分,得到上述Score值的概率的大小.E值越小表示随机情况下得到该Score值的可能性越低.
3.2通过BLAST推测位置蛋白的功能
假如在一次实验中获得了一个蛋白质分子,测定其序列如下:
>unknown protein
MSDKIIHLTDDSFDTDVIKADLAILVDF
WAEWCGPCKMVAPILDEIA
DEFQGKLTVAKLNIDQNPDTAPKYGIRGIPTLLLFKNGEVAANTVGALSKGQLKEFLDANLS
通过BLAST搜索数据库,寻找与之相似的蛋白质序列(图5~图11):1)选择BLAST程序; 2) 输入序列; 3) 选择需要搜索的数据库; 4)选择BLAST程序并运行; 5) BLAST结果显示.
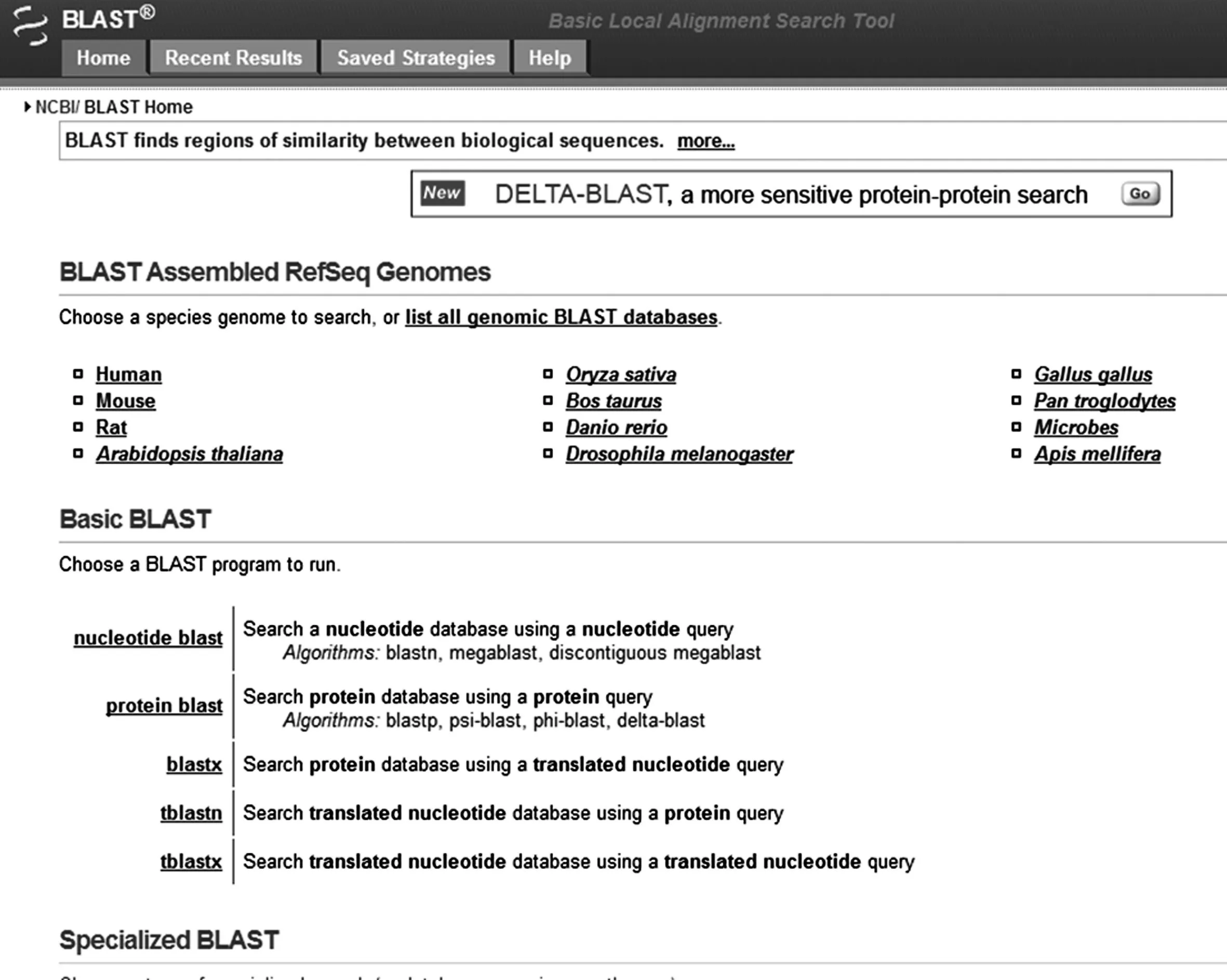
图5 BLAST主界面Fig.5 Main interface of BLAST
图6 序列输入界面Fig.6 Interface of entering query sequence
图7 数据库选择界面Fig.7 Interface of selecting database
图8 程序选择界面Fig.8 Interface of selecting program
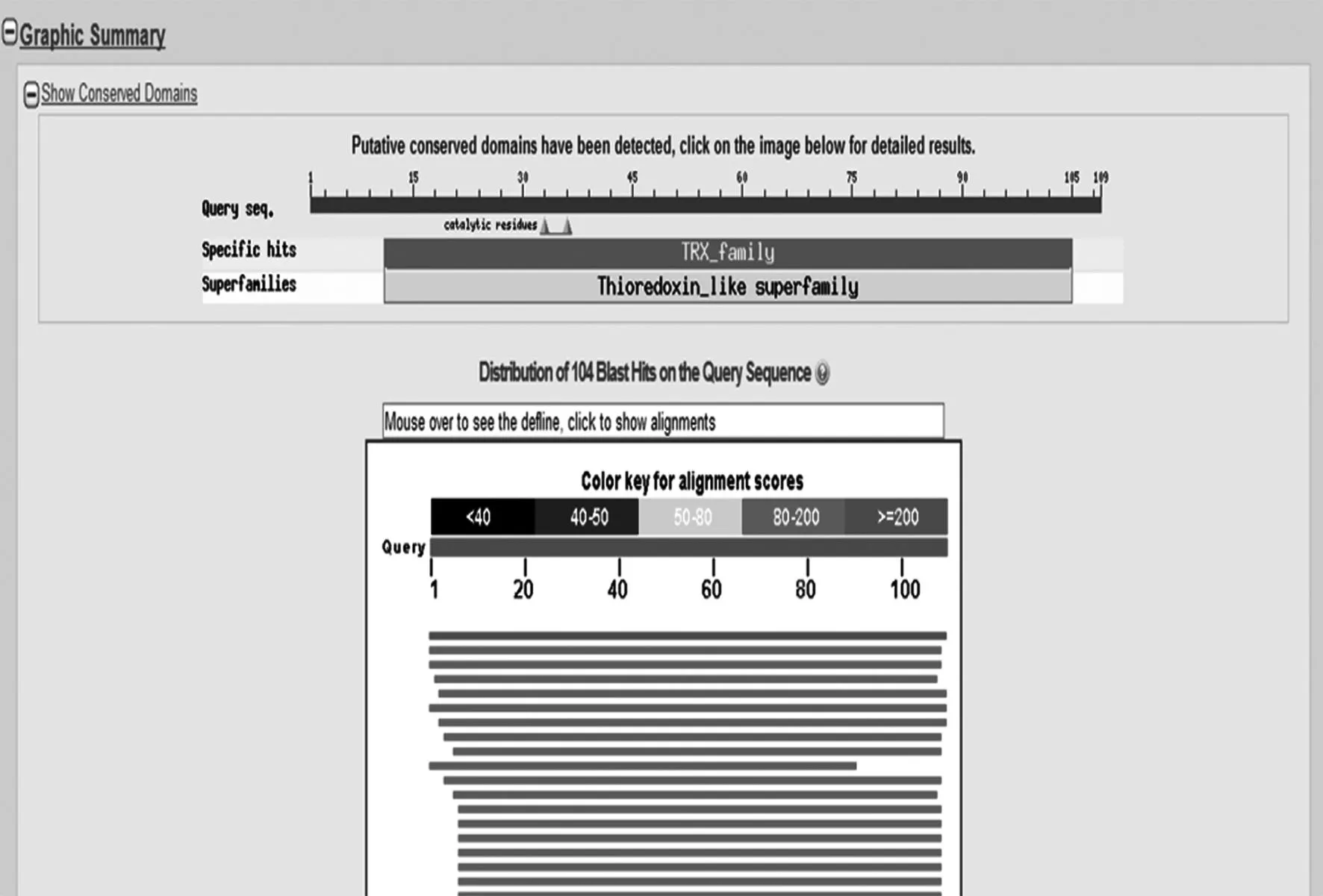
图9 图形表示的结果Fig.9 Graphical result
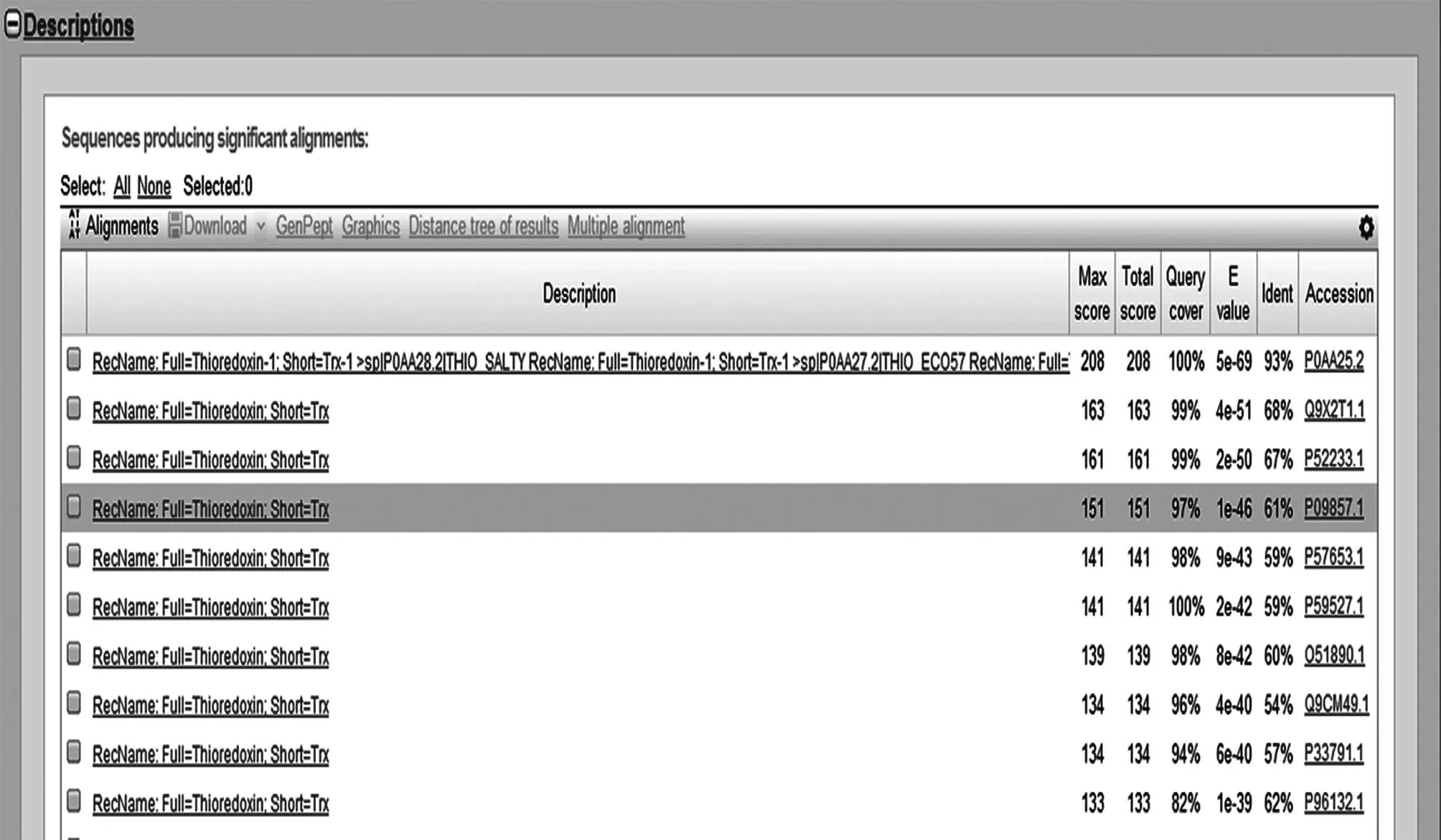
图10 搜索到的序列及信息Fig.10 Sequences and their information
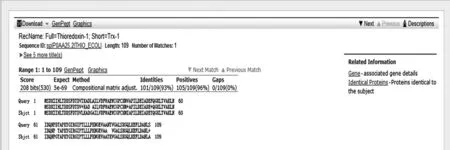
图11 序列比对Fig.11 Sequence alignment
从BLAST搜索结果中发现,未知蛋白与Thioredoxin序列相似性最高,搜索到的蛋白质几乎都是Thioredoxin.因此我们认为未知蛋白为Thioredoxin.其与Escherichia coli K-12的相似性最高(Identities=93%),而与其他物种的相似性都较低( Identities<70% ),因此我们认为未知蛋白可能来自于大肠杆菌,或近似的物种.
3.3利用MEGA软件制作进化树
3.3.1进化树
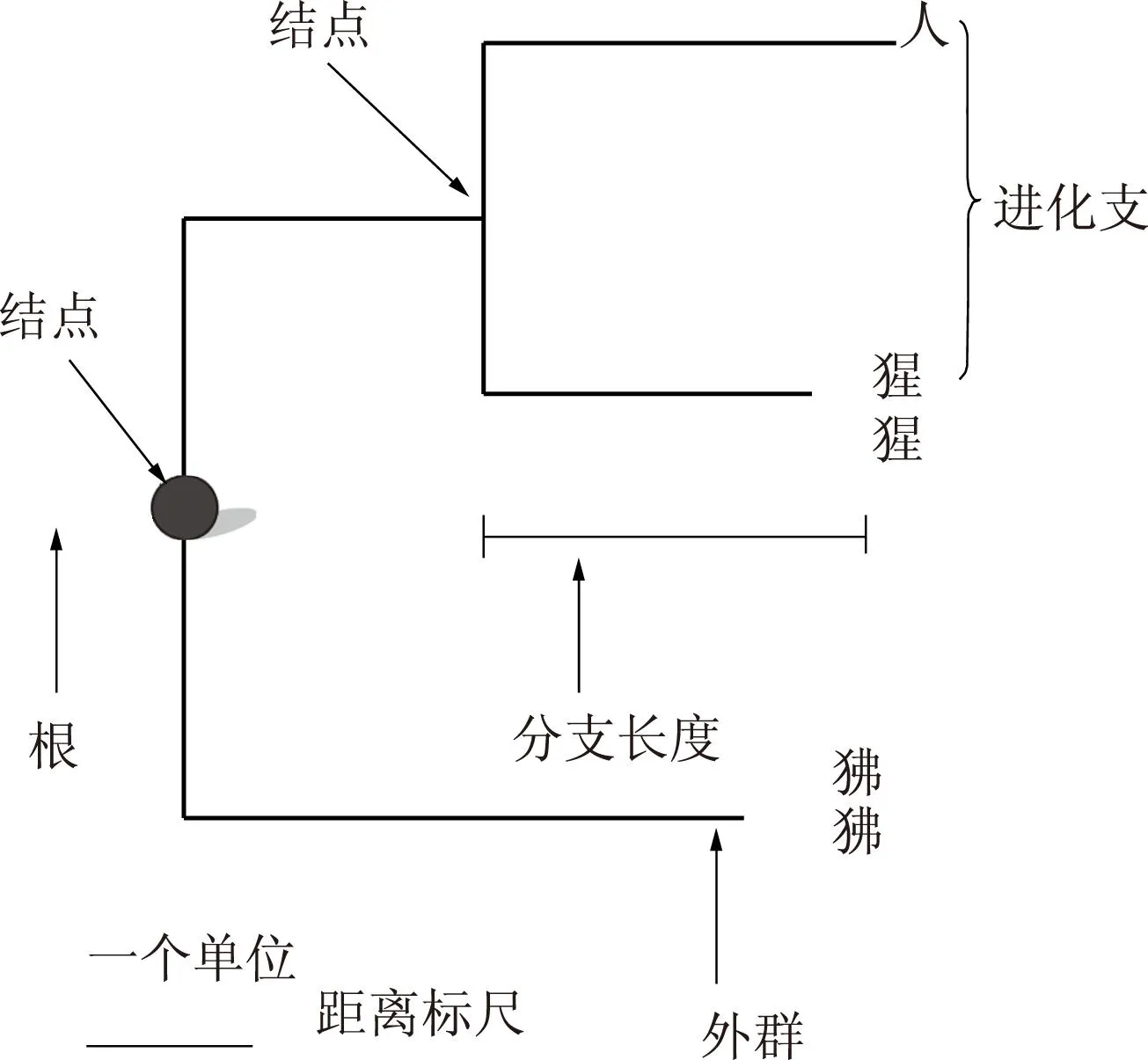
图12 进化树Fig.12 Phylogenetic tree
系统发育进化树(Phylogenetic tree)是用一种类似树状分支的图形概括各种生物间的亲缘关系的图表.进化树由结点(node)和进化分支(branch)组成,每一结点表示一个分类学单元(属、种群、个体等),进化分支定义了分类单元(祖先与后代)之间的关系,一个分支只能连接两个相邻的结点.进化树分支的图像称为进化的拓扑结构,其中分支长度表示该分支进化过程中变化的程度,标有分支长度的进化分支叫标度支(scaled branch).校正后的标度树(scaled tree)常常用年代表示,这样的树通常根据某一或部分基因的理论分析而得出.进化分支可以没有分支长度的标注(unscaled),没有被标注的分支其长度不表示变化的程度,虽然分支的有些地方用数点进行了注释.
进化树可以是有根的(rooted),也可以是无根的(unrooted),分为“有根树”和“无根树”两类.在有根树中,有一个叫根(root)的特殊结点,用来表示共同的祖先,由该点通过唯一途径可产生其他结点;有根树是具有方向的树,包含唯一的根结点,没有确认共同祖先或进化途径(图12).最常用的确定树根的方法是使用一个或多个无可争议的同源物种作为“外群”(outgroup),这个外群要足够近,以提供足够的信息,但又不能太近,以免和树中的种类相混.把有根树去掉根即成为无根树.一棵无根树在没有其他信息(外群)或假设(如假设最大支长为根)时不能确定其树根.无根树是没有方向的,向两个演化方向都有可能.
进化树的构建步骤为:获取序列,多序列比对,选择建树方法,建立进化树,评估.
3.3.2MEGA软件
MEGA软件是一款功能强大的分子进化遗传分析软件,尤其是在计算遗传距离、构建分子系统树方面.它包含许多统计学和遗传学算法,其支持的文件格式很多,而且可以直接从测序图谱中读取序列(图13).利用MEGA软件可以对多个序列进行比对,绘制系统发育进化树,同时可以根据未知物种的基因序列对它的大致分类进行预测.从GenBank等数据库下载基因序列并导入MEGA软件,通过比对可将序列对齐,便于进一步分析. 在程序主界面,点击Analysis->Phylogeny,选择Neighbor-Joining法构建进化树;设置好参数后即可得到进化树(图14).
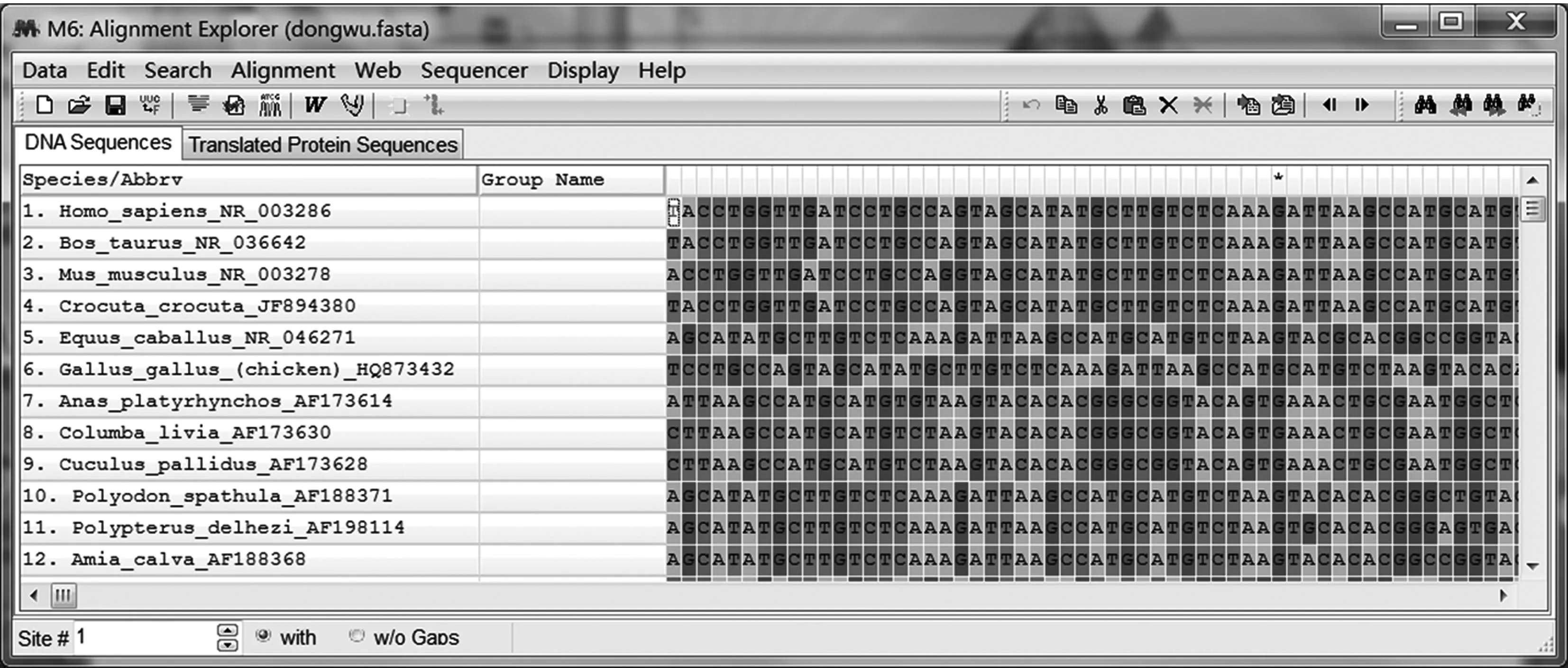
图13 MEGA界面Fig.13 Interface of MEGA
4结论
生物信息学的研究重点主要体现在基因组学和蛋白质学两方面,具体地说就是从核酸和蛋白质序列出发, 分析序列中表达结构和功能的生物信息,并以此为基础预测未知序列的结构与功能.
本文从序列比对的基本概念出发,围绕基本性质阐述序列比对的原理,并通过列举序列比对中常用到的两大工具:基因数据库和计算机软件,说明序列比对在当今生物学研究中起到的重要作用[4].
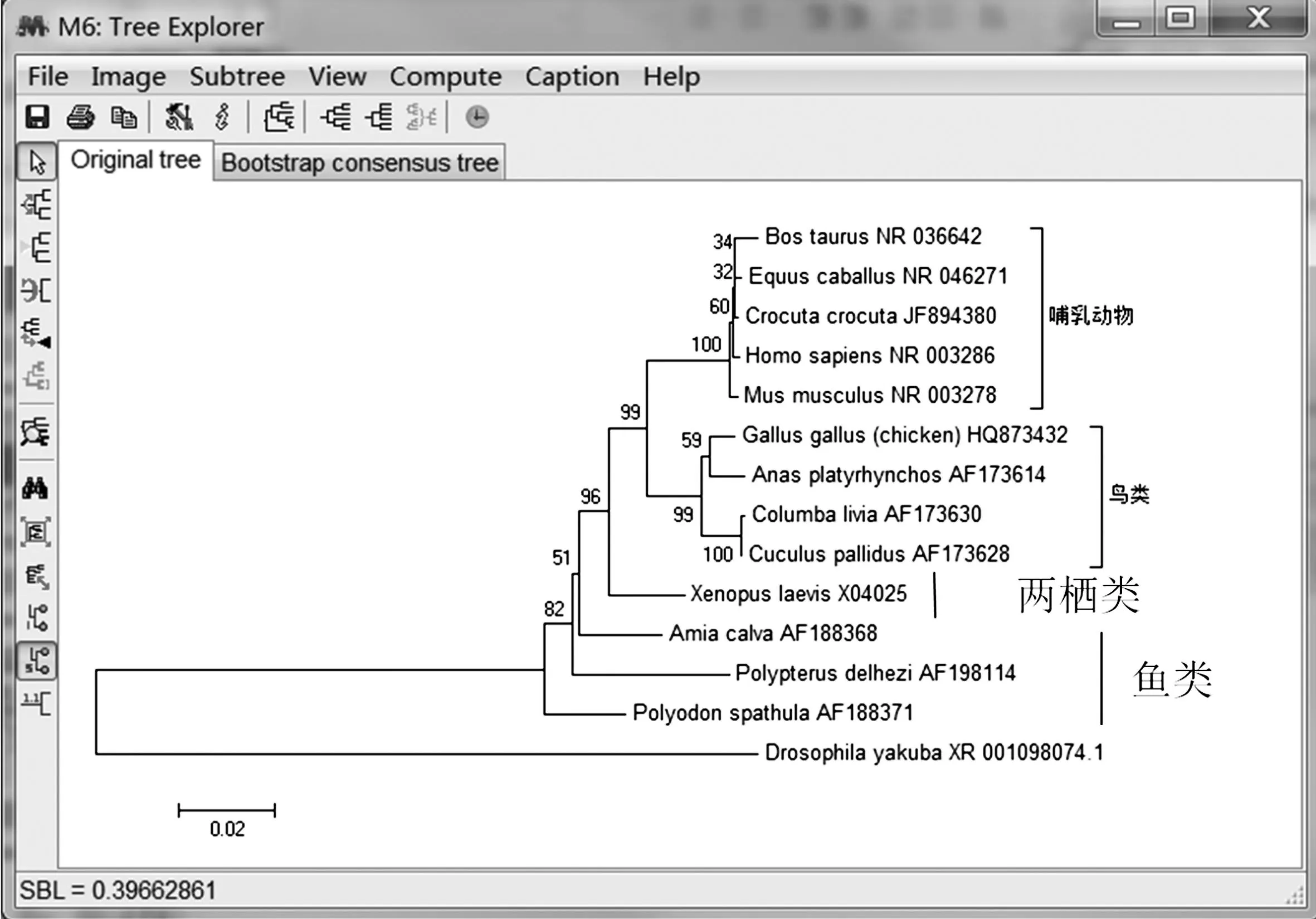
图14 分析结果Fig.14 Result of analysis
[1]ATTWOOD T K , PARRY-SMITH D J.生物信息学概论[M]. 罗静初,译.北京:北京大学出版社,2001:141-145.
[2]DAVID R POWELL, LLOYD ALLISON, TREVOR I DIX. A versatile divide and conquer technique for optimal string alignment[J]. Information Processing Letters, 1999;70:129-139.
[3]ALTSCHUL S F, CISH W M, W MYER E W, et al. Basic local alignment search tool[J]. Journal of Molecular Biology,1990,215:403-410.
[4]HAN JIAWEI, MICHELINE K.数据挖掘概念与技术[M]. 范明,孟晓峰,译.北京:机械工业出版社,2001:301-302.
Search and Similarity Comparison of Gene Sequence
RU Jiakang1, YUAN Lin2
(1.InstituteofLifeSciences,LiaoningUniversity,Shenyang110036,China;2.DepartmentofPharmacy,People’sHospitalofHenanProvince,Zhengzhou450003,China)
Abstract:The basic task of bioinformatics is to analyze all kinds of biological sequences, that is, to study new methods using computers to obtain the knowledge of gene structure, function and evolution from a large amount of sequence information and store it in the database. In sequence analysis, it is a powerful study method to compare the similarity between the unknown sequence and the known sequence in the gene database, including fragment determination, splicing and gene expression analysis as well as the prediction of structure functions of RNA and protein.
Key words:sequence alignment; analysis; prediction
中图分类号:Q31
文献标志码:A
文章编号:1007-0834(2016)01-0025-07
doi:10.3969/j.issn.1007-0834.2016.01.007
作者简介:茹家康(1994—),男,河南郑州人,辽宁大学生命科学院.
基金项目:河南省科技发展计划项目(142102310406)
收稿日期:2015-10-16
