有机半导体Terazulene单晶双极电荷传输性质的理论研究
2016-04-08陈九菊
陈九菊
(黑龙江工程学院电气与信息工程学院, 哈尔滨 150050)
有机半导体Terazulene单晶双极电荷传输性质的理论研究
陈九菊
(黑龙江工程学院电气与信息工程学院, 哈尔滨 150050)
摘要利用密度泛函方法计算Terazulene单晶的重组能和分子间电子耦合, 结合Marcus电荷转移速率理论以及随机行走技术模拟电荷迁移率, 分别研究Terazulene单晶中电子与空穴的角分辨各向异性迁移率及平均迁移率. 结果表明, Terazulene单晶具有均衡的电子与空穴传输性质, 并分析了具体原因. 由于p型有机半导体Naphthodithiophene(NDT)的分子共轭长度与Terazulene接近, 通过比较Terazulene单晶和NDT单晶中电荷传输的差异, 从理论上理解分子结构对有机半导体材料电荷传输性能的影响.
关键词有机半导体Terazulene; 电荷迁移率; 量子化学计算; 电荷转移速率; 随机行走模拟
在有机光电子器件(如有机发光二极管[1]、有机场效应晶体管[2]及有机太阳能电池[3])中, 载流子传输是一个极其重要的物理过程, 载流子传输效率是衡量有机光电子器件性能的一个重要指标. 由于有机分子具有空间各向异性, 所以在固态聚集状态下的有机分子结构以及分子取向对电荷传输有重要影响[4]. 理解有机半导体中电荷传输性能与结构之间的关系, 对于设计合成新材料从而改进器件性能具有重要意义.
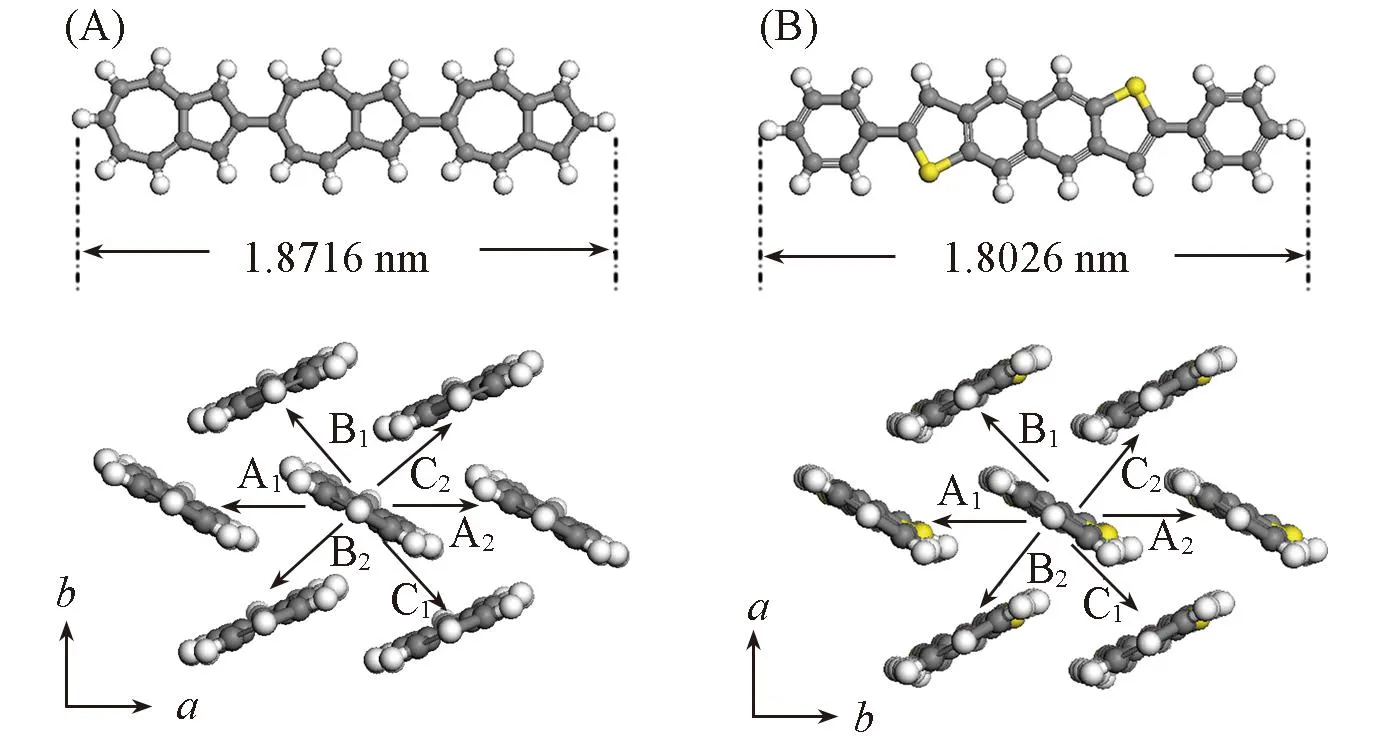
Fig.1 Molecular structures of terazulene(A) and NDT(B) as well as the molecular packing in their ab planes
传统的碳氢化合物(如并五苯)通常具有p型传输性质[5,6], 其近邻分子的最高占据分子轨道(HOMO)间相互作用通常比最低非占据分子轨道(LUMO)间相互作用大[7], 从而有机半导体中的空穴迁移率通常大于电子迁移率. 同时, 由于HOMO能级比LUMO能级更低, 因此p型有机半导体材料具有更好的空气稳定性. 然而, 最近的实验结果表明, 一种有机半导体碳氢化合物Terazulene表现出优越的电子传输性能, 但缺乏空穴传输性能[8]. 因此从理论上研究该有机半导体材料中的载流子传输性质, 对于实验设计新型有机半导体材料具有指导意义. 同时, p型有机半导体材料Naphthodithiophene(NDT)[9]的分子共轭长度与Terazulene相似(结构见图1). 本文从理论上对比研究Terazulene和NDT单晶中本征电子和空穴传输, 有助于理解terazulene分子结构对电荷传输性能的影响.
1计算方法
1.1理论模型
通常有2种模型用于计算有机半导体中的电荷传输: (1) 能带模型[10]; (2) 跳跃模型[11,12]. 因为有机分子通过弱相互作用聚集在一起, 能带模型通常适用于低温下具有周期性结构的有机材料. 在室温下, 有机半导体具有明显的热涨落性质[13], 电荷局域在一个或几个分子上, 跳跃模型可以合理预测电荷迁移率的趋势[14]以及电荷迁移率随温度的变化关系[15,16]. 因此, 本文采用跳跃模型模拟图1中体系的电荷传输. 电荷跳跃速率通过半经典的Marcus电荷转移速率计算[17]:

(1)
式中:V为2个分子间的转移积分(或者电荷耦合); λ为重组能: kB为玻尔兹曼常数;T为温度; ΔG为电荷转移前后的自由能差; ћ=h/2π,h为普朗克常数. 对于自交换反应, ΔG=0.
给定电荷转移速率, 电荷迁移率可通过爱因斯坦[18]关系来计算:

(2)
式中:e为电子的电量;D为扩散系数, 通常表示为电荷扩散距离的方差r2与时间t的比值:
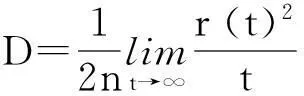
(3)
式中:n表示电荷传输的维度. 图1中2种体系的晶体沿着c轴具有明确的层状结构, 层与层之间相互作用对ab平面内电荷传输的影响很小, 因此本文计算ab平面内的电荷传输, 即n=2. 角分辨的扩散系数可表示为

(4)
式中:n=1, α为相对于ab平面内参考轴的角度. 将式(4)带入式(2), 可以得到角分辨各向异性迁移率.
1.2计算细节
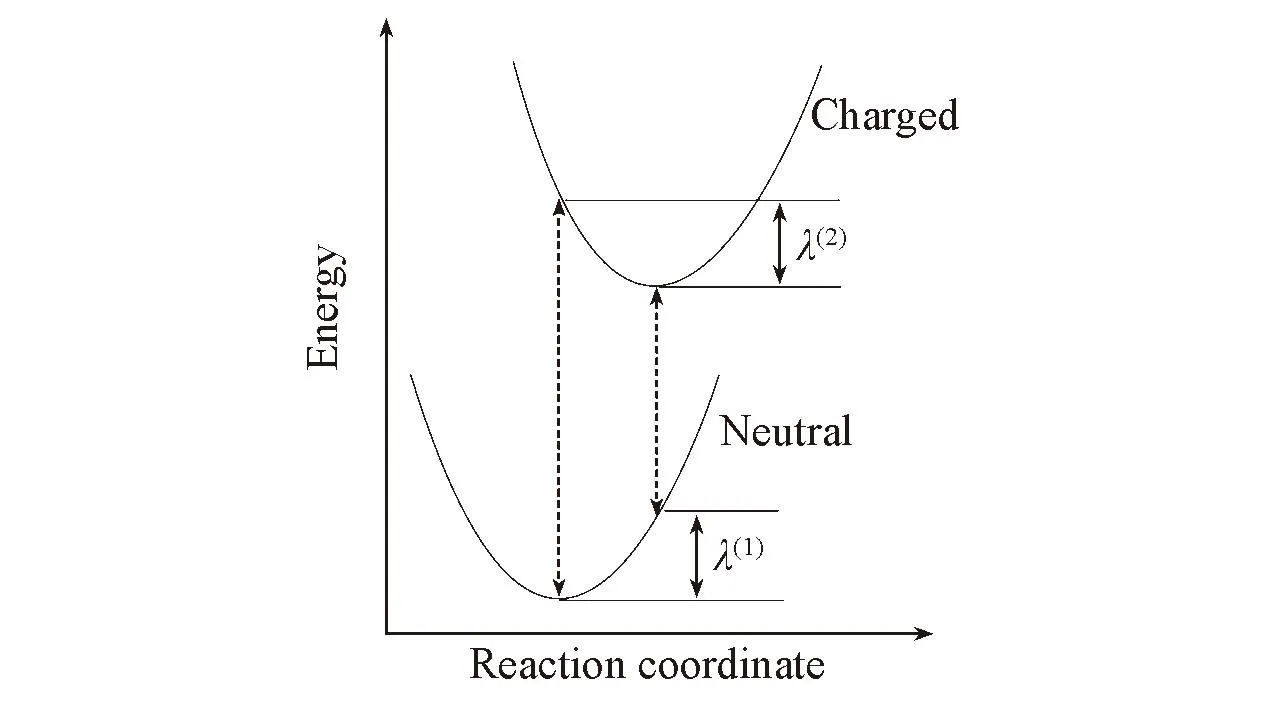
Fig.2 Sketch of the molecular adiabatic potential energy surfaces for the neutral and charged statesλ(1) andλ(2) are the relaxation energies.
由于环境引起的重组能远远小于分子内部的重组能[19], 故本文只计算分子内部重组能, 其由两部分组成, 即λ=λ(1)+λ(2). 由图2可知, λ(1)为中性分子在其弛豫构型和弛豫离子构型下的能量差; λ(2)为离子态分子在其弛豫构型和弛豫中性构型下的能量差. 本文中, λ根据绝热势能面方法[20,21], 利用密度泛函理论(DFT)方法, 选取B3LYP泛函和6-31+G(d,p)基组计算得到.
根据实验得到晶体结构, 选取所有可能的分子对. 每一个分子对间的转移积分根据格点能修正的方法得到[22], 其表达式为

(5)
式中: ei=〈Φi|H|Φi〉(i=1, 2); h12=〈Φ1|H|Φ2〉; S12=〈Φ1|S|Φ2〉.H和S分别为分子对的哈密顿量和重叠矩阵. 对于空穴转移, Φ1和Φ2分别为分子对中2个分子的HOMOs; 对于电子转移, Φ1和Φ2分别为分子对中2个分子的LUMOs. 本文利用DFT方法,分别选用PW91PW91泛函和B3LYP泛函, 并采用6-31+G(d,p)基组计算转移积分. 所有量子化学计算均采用Gaussian 03程序完成[23].

Fig.3 Squared displacement (t) vs. simulation time in the ab plane of terazulene single crystal at 300 K

2结果与讨论
由式(1)可见, 重组能和转移积分是决定有机半导体中电荷传输的2个关键参数. 较小的重组能和较大的转移积分有利于电荷传输. 计算得到Terazulene的空穴重组能和电子重组能分别为151和181 meV, 二者比较接近, 表明重组能对于Terazulene单晶中的空穴传输和电子传输起着相似的作用. 然而, 对于NDT, 其空穴重组能和电子重组能分别为148和288 meV, 可见, 前者远远小于后者. 因此从重组能方面考虑, NDT单晶更有利于空穴传输.
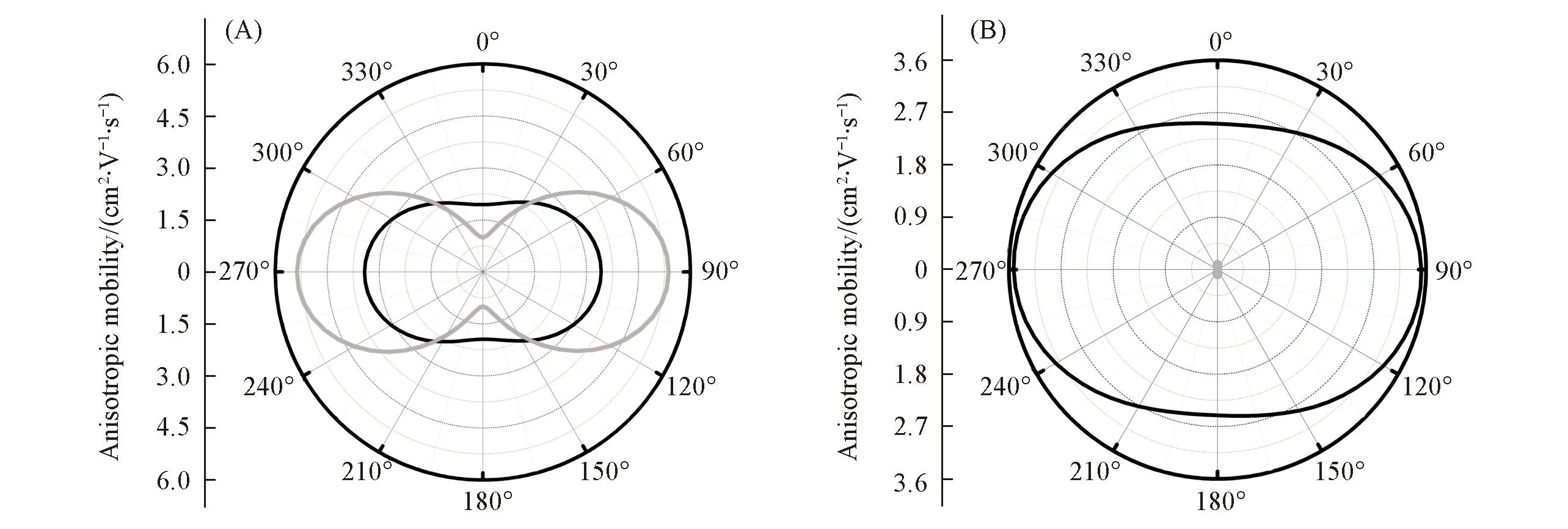
Fig.4 Angular resolution anisotropic mobilities for hole and electron transport in the ab plane of Terazulene(A) and NDT(B) single crystals as a function of αBlack lines represent the hole mobilities and gray lines represent the electron mobilities.
分子间电子耦合与分子间相对位置有密切关系. 为了描述实验中分子间电子耦合, 本文计算的转移积分都是根据实验得到的晶体结构. 由图1可见, 对于Terazulene单晶结构, 其ab平面内每个分子周围有6个最邻近的分子, 每个分子对的转移积分示于表1. 通过对比不同泛函得到的转移积分可以看到, 改变泛函对空穴转移积分和电子转移积分的影响都较小, 所以基于PW91PW91泛函得到的转移积分被用于模拟Terazulene中的载流子迁移率. 对于电子传输, 分子对A1和A2具有最大的转移积分, 而分子对B1和B2具有最小的转移积分. 分子对A1和A2的转移积分大约是分子对B1和B2转移积分的3倍. 图4(A)表明, Terazulene单晶中沿着a轴方向的电子迁移率大于5.0 cm2·V-1·s-1, 而沿着b轴方向的电子迁移率约等于1.0 cm2·V-1·s-1, 所以, 电子迁移率在Terazulene单晶的ab平面内表现出强烈的各向异性. 然而, 对于空穴传输, 实验上并未测量出空穴传输性质[8], 但实验指出邻近2个分子的HOMO轨道劈裂可达到290 meV, 表明空穴转移积分是可以与本文的计算结果(见表1)进行较的. 计算表明, 转移积分在ab平面的不同方向更加均衡, 所以空穴迁移率在Terazulene单晶的ab平面

Table 1 Intermolecular transfer integrals of holes(Vh) and electrons(Ve) for the pathways of
Table 22D Average mobility of holes and electrons in theabplane of terazulene and NDT single crystals, respectively

Species Averagemobility/(cm2·V-1·s-1)TerazuleneCentrosymmetricNDTHole2.67±0.103.09±0.09Electron3.08±0.090.064±0.004
内表现出较弱的各向异性[见图4(A)]. 比较图4(A)中的空穴迁移率和电子迁移率可见, 沿着a轴方向, 电子迁移率比空穴迁移率大, 而沿着b轴方向, 电子迁移率比空穴迁移率小. 当把空穴和电子的各向异性迁移率进行平均后, 从表2可见, Terazulene单晶中ab平面内平均空穴传输能力与平均电子传输能力相似. 计算表明, 重组能和转移积分对于空穴传输和电子传输都比较接近, 表明Terazulene是一种双极传输材料. NDT的分子共轭长度与Terazulene相近, 其转移积分列于表1. 可见, NDT的空穴转移积分在A1和A2方向与电子转移积分相似, 但在B1, B2, C1和C2方向远远大于电子转移积分. 同时, 考虑到NDT的空穴重组能明显小于电子重组能, 在NDT单晶的ab平面各个方向, 空穴迁移率都远远大于电子迁移率[图4(B)], 从而ab平面内的平均空穴传输能力远远大于平均电子传输能力(表2). 因此, NDT的重组能和转移积分都有利于空穴传输, 使得NDT表现出空穴传输性质.
3结论
利用DFT方法、Marcus电荷转移速率理论以及随机行走技术, 从理论上研究了Terazulene单晶中的电荷传输, 并系统考察了重组能和转移积分对Terazulene单晶中电荷传输的影响. 研究表明, 空穴和电子的重组能比较接近, 空穴转移积分分布在电子转移积分的范围内. 因此, 与实验测量不同的是, 研究预测Terazulene是一种双极电荷传输材料. 本文推测实验在将Terazulene制备成场效应晶体管器件时, 在Terazulene与衬底的界面附近引入了限制空穴传输的缺陷. 同时, 研究表明NDT是一种空穴传输材料, 证实了实验的测量结果. 由于NDT的分子共轭长度以及固态聚集方式与Terazulene相似, 所以NDT与Terazulene不同的电荷传输性质归结于分子结构的变化.
参考文献
[1]Farinola G. M., Ragni R.,Chem.Soc.Rev., 2011, 40, 3467—3482
[2]Mas-Torrent M., Rovira C.,Chem.Rev., 2011, 111(8), 4833—4856
[3]Clarke T. M., Durrant J. R.,Chem.Rev., 2010, 110(11), 6736—6767
[4]Brédas J. L., Beljonne D., Coropceanu V., Cornil J.,Chem.Rev., 2004, 104(11), 4971—5003
[5]Jurchescu O. D., Baas J., Palstra T. T. M.,Appl.Phys.Lett., 2004, 84(16), 3061—3063
[6]Sundar V. C., Zaumseil J., Podzorov V., Menard E., Willett R. L., Someya T., Gershenson M. E., Rogers J. A.,Science, 2004, 303, 1644—1646
[7]Shirota Y., Kageyama H.,Chem.Rev., 2007, 107(4), 953—1010
[8]Yamaguchi Y., Ogawa K., Nakayama K., Ohba Y., Katagiri H.,J.Am.Chem.Soc., 2013, 135, 19095—19098
[9]Shinamura S., Osaka I., Miyazaki E., Nakao A., Yamagishi M., Takeya J., Takimiya K.,J.Am.Chem.Soc., 2011, 133, 5024—5035
[10]Cheng Y. C., Silbey R. J., da Silva Filho D. A., Calbert J. P., Cornil J., Brédas J. L.,J.Chem.Phys., 2003, 118(8), 3764—3774
[11]Hutchison G. R., Ratner M. A., Marks T. J.,J.Am.Chem.Soc., 2005, 127(7), 2339—2350
[12]Deng W. Q., Goddard Ⅲ W. A.,J.Phys.Chem.B, 2004, 108(25), 8614—8621
[13]Troisi A., Orlandi G.,J.Phys.Chem.A, 2006, 110(11), 4065—4070
[14]Song Y. B, Di C. A., Yang X. D., Li S. P., Xu W., Liu Y. Q., Yang L. M., Shuai Z. G., Zhang D. Q., Zhu D. B.,J.Am.Chem.Soc., 2006, 128(50), 15940—15941
[15]Nan G. J., Yang X. D., Wang L. J., Shuai Z. G., Zhao Y.,Phys.Rev.B, 2009, 79(11), 115203
[16]Geng H., Peng Q., Wang L. J., Li H. J., Liao Y., Ma Z. Y., Shuai Z. G.,Adv.Mater., 2014, 24, 3568—3572
[17]Marcus R. A.,Rev.Mod.Phys., 1993, 65(3), 599—610
[18]Schein L. B., McGhie A. R.,Phys.Rev.B, 1979, 20, 1631—1639
[19]Norton J. E., Brédas J. L.,J.Am.Chem.Soc., 2008, 130(37), 12377—12384
[20]Malagoli M., Brédas J. L.,Chem.Phys.Lett., 2000, 327, 13—17
[21]Wen S. H., Deng W. Q., Han K. L.,Phys.Chem.Chem.Phys., 2010, 12, 9267—9275
[22]Valeev E. F., Coropceanu V., da Silva Filho D. A., Salman S., Brédas J. L.,J.Am.Chem.Soc., 2006, 128(30), 9882—9886
[23]Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Montgomery J. A., Vreven T., Kudin K. N., Burant J. C., Millam J. M., Iyengar S. S., Tomasi J., Barone V., Mennucci B., Cossi M., Scalmani G., Rega N., Petersson G. A., Nakatsuji H., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Klene M., Li X., Knox J. E., Hratchian H. P., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Ayala P. Y., Morokuma K., Voth G. A., Salvador P., Dannenberg J. J., Zakrzewski V. G., Dapprich S., Daniels A. D., Strain M. C., Farkas O., Malick D. K., Rabuck A. D., Raghavachari K., Foresman J. B., Ortiz J. V., Cui Q., Baboul A. G., Clifford S., Cioslowski J., Stefanov B. B., Liu G., Liashenko A., Piskorz P., Komaromi I., Martin R. L., Fox D. J., Keith T., Al-Laham M. A., Peng C. Y., Nanayakkara A., Challacombe M., Gill P. M. W., Johnson B., Chen W., Wong M. W., Gonzalez C., Pople J. A., Gaussian 03, Revision E.01, Gaussian Inc., Wallingford CT, 2004
[24]Yang X. D., Wang L. J., Wang C. L., Long W., Shuai Z. G.,Chem.Mater., 2008, 20(9), 3205—3211
Theoretical Studies on the of Ambipolar Charge
Transport in Terazulene Single Crystal†
CHEN Jiuju*
(CollegeofElectricalandInformationEngineering,
HeilongjiangInstituteofTechnology,Harbin150050,China)
AbstractThe quantum chemical calculations and the Marcus charge transfer theory were combined to study the charge transport property for holes and electrons in the terazulene single crystal. The angular resolution anisotropic and average charge mobilities were obtained simultaneously from a set of identical trajectories with random walk technique. Meanwhile, Terazulene showed similar molecular conjugation to naphthodithiophene, which was a good hole transport material. The different types of charge transport for terazulene and naphthodithiophene were analyzed, which provided insight for the influence of the molecular structures on the charge transport from a theoretical viewpoint.
KeywordsOrganic semiconductor terazulene; Charge mobility; Quantum chemical calculation; Charge transfer rate; Random walk simulation
(Ed.: Y, Z)
† Supported by the Youth Science Foundation of Heilongjiang Institute of Technology, China(No.2014QJ10).
doi:10.7503/cjcu20150442
基金项目:黑龙江工程学院青年科学基金项目(批准号: 2014QJ10)资助.
收稿日期:2015-06-08. 网络出版日期: 2015-12-20.
中图分类号O649
文献标志码A
联系人简介: 陈九菊, 女, 讲师, 主要从事半导体器件的研究. E-mail: jiujuchen09@163.com
