内脏异位综合征的研究进展
2016-04-01孙妍王剑鹏李慧李晓妮张茗卉张丽综述王浩审校
孙妍、王剑鹏、李慧、李晓妮、张茗卉、张丽综述,王浩审校
内脏异位综合征的研究进展
孙妍、王剑鹏、李慧、李晓妮、张茗卉、张丽综述,王浩审校
摘要内脏异位综合征的根本原因是侧分化异常。该病主要累及消化系统、呼吸系统以及循环系统等多个系统。内脏异位综合征是一类因胚胎期内脏左右侧非对称结构的无法正常建立而导致的综合征,可分左侧异构及右侧异构。研究发现大多数内脏异位患者存在原发纤毛运动障碍。随着对该病认识和检测方法的提高,越来越多的内脏异位综合征被早期甚至胎儿期确诊。左侧异构患者一般可行双心矫治,右侧异构常包括肺静脉异位引流、体静脉异位引流、单心房、单心室、共同房室瓣等复杂畸形,大多数患者难以达到解剖矫治,只能采取分阶段姑息和功能矫治手术。目前内脏异位综合征并发心脏畸形的治疗效果并不理想,我们期待通过分子生物学的深入研究寻找致病机制。
关键词综述;内脏异位;心脏缺损,先天性
内脏异位(Heterotaxy)是一组广泛累及心脏及众多心外器官的综合畸形,遗传学研究表明其发生与胚胎早期发育异常密切相关。2000年,国际儿童和先天性心脏病的命名委员会成立。他们对“Heterotaxy”的定义为内脏异位综合征,表现为胸腹部脏器沿身体左右轴异常排列。内脏异位并不包括内部器官沿左右轴正常排列,即“内脏正位”,也不包括脏器完全沿左右轴反位排列的镜像患者,即“内脏反位(镜面)”。这类疾病按照其排列形态又被分为左侧异构及右侧异构。由于两种异构类型各自独特的心耳特点被分别称双侧左心耳结构或左心房异构(表现为两个心耳均呈左心耳样,基底较窄、手指形)及双侧右心耳结构或左心房异构(表现为两个心耳均呈右心耳样,基底较宽、三角形)[1,2],也有研究者根据脾脏的状态将内脏异位分为多脾综合征(左侧异构)和无脾综合征(右侧异构)[3-5]。
1 内脏异位综合征的病理学特征
先天性心脏病发病率占新生儿的0.75%~0.9%[6, 7],也是新生儿的首要死因。内脏异位综合征的发病率占先天性心脏的1/7000~1/5000[8, 9]。其常见心血管畸形谱见表1。除心血管系统异常外,内脏异位综合征还伴有呼吸及循环等系统的一系列特征性异常改变(表2)。
相对于左侧异构患者,右侧异构患者心脏畸形更为复杂且严重。国内一项对无脾患儿的研究[10]共入组47例患儿,其中男性27例(57.4%),女性20例(42.6%)。研究者将患儿所存在的各类心脏畸形按出现频次进行频数统计。结果表明频率较高的类别包括心脏位置异常(34%,包括右位心、中位心等)、大范围间隔组织缺损(100%,包括单心房、单心室、房室间隔缺损)、房室瓣异常(89.4%,包括共同房室瓣,单组房室瓣或二、三尖瓣闭锁)、主动脉起源异常(100%,包括右心室双出口、大动脉转位)、大动脉相互关系异常(97.8%,除主动脉位于肺动脉右后方外的其他类型)、右心室流出道、肺动脉瓣及肺动脉发育异常(95.7%,包括右心室流出道狭窄、闭锁,肺动脉瓣狭窄、闭锁,主肺动脉及左右分支发育不良)、体静脉引流异常(91.5%,包括包括肝静脉直接入左心房、腔静脉异位引流)、肺静脉引流异常(59.6%,包括完全或部分肺静脉异位引流)。王剑鹏等[10]总结无脾患者静脉引流特点:(1)存在双侧上腔静脉者占61%;(2)双侧上腔均引流入双侧心房或单心房两侧;(3)肝静脉数目常多于3支,约半数直接汇入心房不同部位;(4)90%患者存在体静脉畸形引流;(5)存在共同肺静脉干者约占45%,存在肺静脉异位引流者约占57.1%。
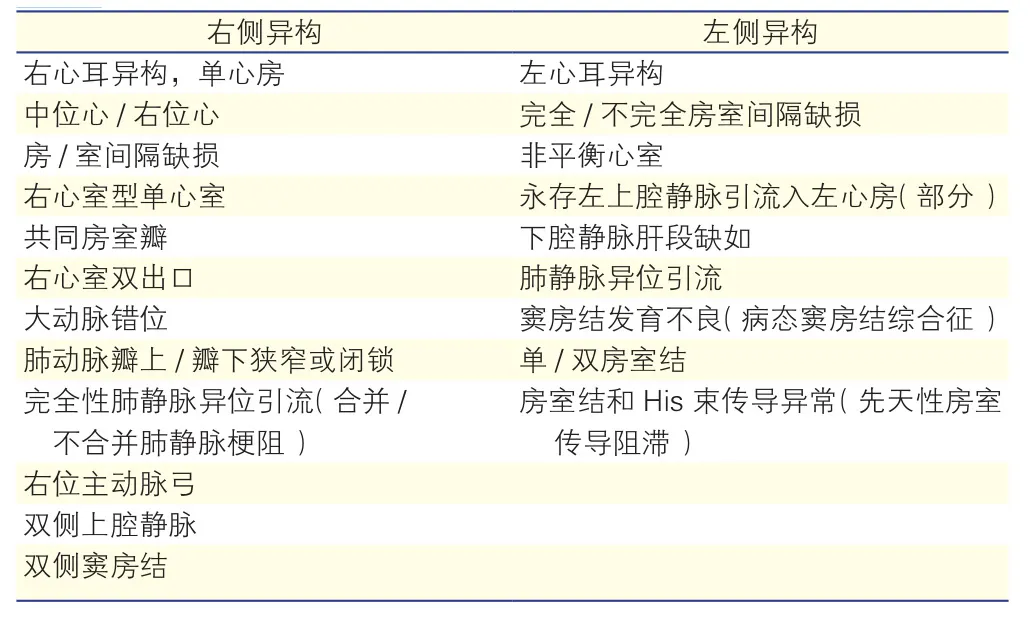
表1 内脏异位综合征的心血管畸形特征
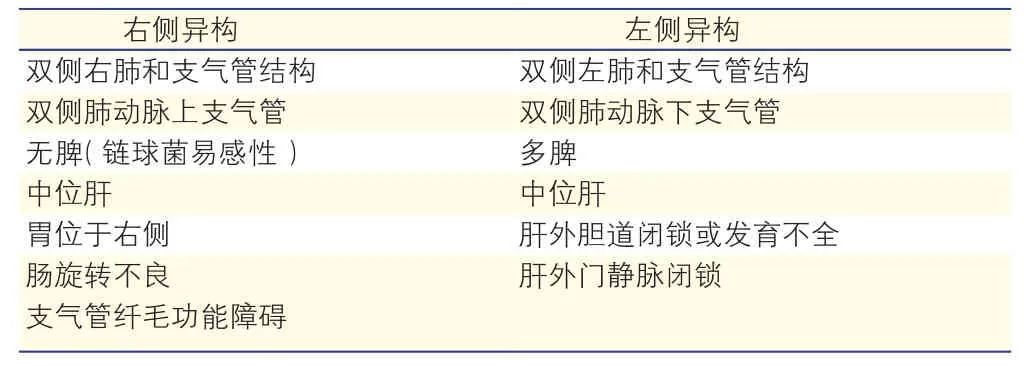
表2 内脏异位综合征的心外畸形特征
2 内脏异位综合征的分子生物学机制
内脏异位综合征是一类因胚胎期内脏左右侧非对称结构的无法正常建立而导致的综合征。研究发现大多数内脏异位患者存在原发纤毛运动障碍(PCD),表现为纤毛运动异常或纤毛无运动[11, 12]。
近年来,动物实验对左右体轴的确定过程的机制进行了广泛的研究。研究者发现胚胎发育最初内脏是对称的,而由于原结上出现的自由向左的“结流”首次打破了对称发育的进程[13]。所谓“结流”是指结细胞纤毛顺时针旋转所产生的液体流,液体流使得原结左侧的钙离子浓度增高,从而产生了不对称的信号,这些信号转而传向左侧板中胚层,从而激发了下游左右特异生长的基因和转录因子[13-15]。
通过其他一系列未知的因子开始调控机体左侧的基因表达,并产生左右不对称的器官发育过程。随着对研究者对基因水平的深入研究,人们发现480个与内脏左右轴排列相关的基因,其中一些基因可能具有保守性,它们的突变已经被证实与内脏异位相关。这些基因包括ZIC3、NODAL、CFC1、ACVR2B、LEFTY2、CITED2和GDF[16-21]。然而这类基因的确切功能和作用机制还有待于人类更深入的研究。
3 内脏异位综合征的临床诊断
过去对内脏异位综合征的诊断主要依赖尸检。随着对该病认识的提高和超声诊断、心血管造影、计算机断层扫描(CT)、核磁共振成像(MRI)等技术的普遍应用,越来越多的内脏异位综合征被早期诊断出来,有的甚至在胎儿期便可获得明确诊断。超声检查对该类疾病诊断有重要价值,不仅可以观察到相对特征性的心脏畸形,也可以观察到腹腔脏器的改变。超声心动图检查除可对心脏解剖进行描述外,还可提供血液动力学改变。对于该类患者,肝脏位置、形态,脾脏的缺如或分叶也必须单独描述,这对临床诊断内脏异位综合征有重要价值。胸部X片如果显示新生儿右位心,心影扩大,肺血减少,主支气管形态及夹角异常多提示存在内脏异位。CT可以清晰地显示缺如或分叶的脾脏,明确其数目及位置,显示肝脏及胆囊、下腔静脉、奇静脉等。戴汝平等[22]认为对于单心室者,CT扫描应包括一部分腹部脏器,以便了解有否存在无脾或多脾综合征。他同时指出:因无脾综合征易并发肺静脉异位连接,故如发现患者无脾,应警惕肺静脉异位连接的存在。CT还可显示主支气管长度及其与中轴的夹角[23, 24],均对该病有提示价值。CT血管造影检查进一步明确心内畸形、肺动脉发育及体肺静脉异位引流途径、有无狭窄、血管连接关系等,为外科制定治疗方案提供证据。MRI可对器官进行多维面的扫描,并能清楚地显示心脏大血管,对显示多脾综合征的各种畸形可能有较大帮助。由于该类疾病出现心脏异常累及各个节段,因而描述此类心脏异常应包括静脉与心房连接、心脏的位置、心房形态、心房心室连接、心室襻、心室形态、心室动脉连接、主干动脉的空间关系以及各瓣膜结构功能等。
4 内脏异位综合征的治疗方法及预后
左侧异构患者一般心内畸形相对较轻,部分患者可行双心室矫治。右侧异构(无脾综合征)常包括肺静脉异位引流、体静脉异位引流、单心房、单心室、共同房室瓣等复杂畸形,大多数患者难以达到解剖矫治,只能采取分阶段姑息和功能矫治手术。在先天性心脏病的诊疗技术大为提高的今天,右侧异构合并心内畸形仍存在较高的病残率和病死率,是目前预后最差的先天性心脏病之一,5年平均生存率为30%~74%[1, 2, 25]。随着胎儿超声心动图检查的日益发展,更多的内脏异位综合征在胚胎期就被检出,然而国外文献显示早期诊断并不能降低内脏异位综合征患儿死亡率。
目前手术治疗方法包括双心室矫治和腔静脉-肺动脉连接,然而绝大多数无脾患者仅能施行不同类型的Glenn或Fontan类手术治疗[26-29],极少数患者可行双心室矫治[30]。内脏异位综合征患者死亡的主要危险因素包括共同房室瓣反流、肺静脉狭窄、缺少肺动脉狭窄、感染等[31]。内脏异位综合征患儿多存在静脉畸形引流情况,其直接影响手术方式以及手术效果[31-34]。Eronen等[35]报道,内脏异位综合征合并肺静脉异位引流者高达88%。Hashmi等[36]报道,30%内脏异位综合征合并肺静脉梗阻。在Glenn手术中,术前必须明确双侧或单侧上腔静脉及其回流部位,双侧上腔静脉可行双侧Glenn手术;下腔静脉回流至心房的部位,肝静脉是否直接回流至心房及其回流的位置,也将影响到全腔静脉-肺动脉手术的可行性及术式选择。肺静脉异位引流的类型及引流部位对手术方式的选择也非常重要,存在完全肺静脉异位引流的患儿往往意味着需要更早干预,手术难度增大,围手术期死亡增加。既往多篇文献曾报道存在肺静脉异位引流的无脾综合征患儿预后很差[34, 36]。有研究发现内脏异位综合征患者平均肺静脉指数明显小于无内脏异位综合征者。同时他们还认为肺静脉内径对完全肺静脉异位引流患者的生存有很强的预测价值。无脾综合征合并心内畸形的患者死亡率非常高,Eronen等[35]回顾了赫尔辛基大学医学院1976年~2010年诊治的32例无脾综合征患者,中位随访时间13.8年的总生存率仅为22%。心脏移植可能是严重心力衰竭、顽固性心律失常、复发的蛋白丢失性肠病患者的唯一治疗选择[37-40]。
目前内脏异位综合征并发心脏畸形的治疗效果并不理想,我们期待通过分子生物学的深入研究寻找致病机制。我国实行优生优育,孕期如果发现内脏异位伴有严重的心内畸形,应该早期咨询小儿外科医生,听取针对性建议。
参考文献
[1] Jacobs JP, Anderson RH, Weinberg PM, et al.The nomenclature, definition and classification of cardiac structures in the setting of heterotaxy.Cardiol Young, 2007, 17(Suppl 2): 1-28.
[2] Cohen MS, Anderson RH, Cohen MI, et al.Controversies, genetics, diagnostic assessment, and outcomes relating to the heterotaxy syndrome.Cardiol Young, 2007, 17(Suppl 2): 29-43.
[3] Bartram U, Wirbelauer J, Speer CP.Heterotaxy syndrome-asplenia and polysplenia as indicators of visceral malposition and complex congenital heart disease.Biol Neonate, 2005, 88: 278-290.
[4] Chandra RS.Biliary atresia and other structural anomalies in the congenital polysplenia syndrome.J Pediatr, 1974, 85: 649-655.
[5] Applegate KE, Goske MJ, Pierce G, et al.Situs revisited: imaging of the heterotaxy syndrome.Radiographics, 1999, 19: 837-852.
[6] Van der Linde D, Konings EE, Slager MA, et al.Birth prevalence of congenital heart disease worldwide: A systematic review and metaanalysis.J Am Coll Cardiol, 2011, 58: 2241-2247.
[7] Hoffman JI, Kaplan S.The incidence of congenital heart disease.J Am Coll Cardiol, 2002, 39: 1890-1900.
[8] Reller MD, Strickland MJ, Riehle-Colarusso T, et al.Prevalence ofcongenital heart defects in metropolitan Atlanta, 1998-2005.J Pediatr, 2008, 153: 807-813.
[9] Lin AE, Ticho BS, Houde K, et al.Heterotaxy: Associated conditions and hospital-based prevalence in newborns.Genet Med, 2000, 2: 157-172.
[10] 王剑鹏, 孙妍, 李慧, 等.无脾综合征患者体肺静脉回流特点分析.中华医学超声杂志(电子版) , 2015, 2: 160-164.
[11] Maisonneuve C, Guilleret I, Vick P, et al.Bicaudal C, a novel regulator of Dvl signaling abutting RNA-processing bodies, controls cilia orientation and leftward flow.Development, 2009, 136: 3019-3030.
[12] Marszalek JR, Ruiz-Lozano P, Roberts E, et al.Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-Ⅱ.Proc Natl Acad Sci USA, 1999, 96: 5043-5048.
[13] Nonaka S, Tanaka Y, Okada Y, et al.Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein.Cell, 1998, 95: 829-837.
[14] McGrath J, Somlo S, Makova S, et al.Two populations of node monocilia initiate left-right asymmetry in the mouse.Cell, 2003, 114: 61-73.
[15] Levin M, Johnson RL, Stern CD, et al.A molecular pathway determining left-right asymmetry in chick embryogenesis.Cell, 1995, 82: 803-814.
[16] Belmont JW, Mohapatra B, Towbin JA, et al.Molecular geneticsof heterotaxy syndromes.Curr Opin Cardiol, 2004, 19: 216-220.
[17] Zhu L, Belmont JW, Ware SM.Genetics of human heterotaxias.Eur J Hum Genet, 2006, 14: 17-25.
[18] Mohapatra B, Casey B, Li H, et al.Identification and functional characterization of NODAL rare variants in heterotaxy and isolated cardiovascular malformations.Am J Hum Genet, 2007, 81: 987-994.
[19] Kaasinen E, Aittomi K, Eronen M, et al.Recessively inherited right atrial isomerism caused by mutations in growth/differentiation factor 1 (GDF1).Hum Mol Genet, 2010, 19: 2747-2753.
[20] Kosaki R, Gebbia M, Kosaki K, et al.Left-right axis malformations associated with mutations in ACVR2B, the gene for human activin receptor typeⅡB.Am J Med Genet, 1999, 82: 70-76.
[21] Kosaki K, Bassi MT, Kosaki R, et al.Characterization and mutation analysis of human LEFTY A and LEFTY B, homologues of murine genes implicated in left-right axis development.Am J Hum Genet, 1999, 64: 712-721.
[22] 戴汝平.心血管病CT诊断学.北京: 人民卫生出版社, 2000.186-187.
[23] Lim JS, McCrindle BW, Smallhorn JF, et al.Clinical features, management, and outcome of children with fetal and postnatal diagnoses of isomerism syndromes.Circulation, 2005, 112: 2454-2461.
[24] Swisher M, Jonas R, Tian X, et al.Increased postoperative and respiratory complications in patients with congenital heart disease associated with heterotaxy.J Thorac Cardiovasc Surg, 2011, 141: 637-644.
[25] Cheung YF, Cheng VYW, Chau AKT, et al.Outcome of infants with right atrial isomerism:Is prognosis better with normal pulmonary venous drainage? Heart, 2002, 87: 146-152.
[26] Bartz PJ, Driscoll DJ, Dearani JA, et al.Early and late results of the modified fontan operation for heterotaxy syndrome 30 years of experience in 142 patients.J Am Coll Cardiol, 2006, 48: 2301-2305.
[27] Serraf A, Bensari N, Houyel L, et al.Surgical management of congenital heart defects associated with heterotaxy syndrome.Eur J Cardiothorac Surg, 2010, 38: 721-727.
[28] Hoashi T, Ichikawa H, Fukushima N, et al.Long-term clinical outcome of atrial isomerism after univentricular repair.J Card Surg, 2009, 24: 19-23.
[29] Ohuchi H, Kagisaki K, Miyazaki A, et al.Impact of the evolution of the Fontan operation on early and late mortality: A single-center experience of 405 patients over 3 decades.Ann Thorac Surg, 2011, 92: 1457-1466.
[30] Masahiro K, Toshikatsu Y, Hideki U, et al.Biventricular Repair for Right Atrial Isomerism.Ann Thorac Surg, 2006, 81: 1808-1816.
[31] Hutter D, Redington AN.The principles of management, and outcomes for patients with functionally univentricular hearts.In: Anderson RH, Baker EJ, Penny DJ, et al.Paediatric cardiology, 3rd edn.Philadelphia: Churchill Livingstone, 2009, 687-696.
[32] Culbertson DB, George BL, Day RW, et al.Factors influencing survival of patients with heterotaxy syndrome undergoing Fontan procedure.J Am Coll Cardiol, 1992, 20: 678-684.
[33] Morales DL, Braud BE, Booth JH, et al.Heterotaxy patients with total anomalous pulmonary venous return: improving surgical results.Ann Thorac Surg, 2006, 82: 1621-1628.
[34] Sadiq M, Stumper O, De Giovanni JV, et al.Management and outcome of infants and children with right atrial isomerism.Heart, 1996, 75: 314-319.
[35] Eronen M, Aittomaki K, Kajantie E.The outcome of patients with right atrial isomerism is poor.Pediatr Cardiol, 2013, 34: 302-307.
[36] Hashmi A, Abu-Sulaiman R, McCrindle B, et al.Management and outcome of right atrial isomerism: A 26-year experience.J Am Coll Cardiol, 1998, 31: 1120-1126.
[37] Kanter KR, Mahle WT, Vincent RN, et al.Heart transplantation in children with a Fontan procedure.Ann Thorac Surg, 2011, 91: 823-829.
[38] Davies RR, Sorabella RA, Yang J, et al.Outcomes after transplantation for “failed” Fontan: A singleinstitution experience.J Thorac Cardiovasc Surg, 2012, 143: 1183-1192.
[39] Gamba A, Merlo M, Fiocchi R, et al.Heart transplantation in patients with previous Fontan operations.J Thorac Cardiovasc Surg, 2004, 127: 555-562.
[40] 欧艳秋, 刘小清, 麦劲壮,等.复杂性先天性心脏病在非活产缺陷儿中的发生状况.中国循环杂志, 2012, 27: 302-304.
(编辑:许菁)
收稿日期:(2015-08-13)
中图分类号:R54
文献标识码:A
文章编号:1000-3614(2016)02-0203-03
doi:10.3969/j.issn.1000-3614.2016.02.025
作者简介:孙妍 主治医师 博士 主要从事心脏超声研究 Email:sunxiaoyan01@sina.com 通讯作者:王浩 Email:hal6112@gmail.com
作者单位:100037 北京市,中国医学科学院 北京协和医学院 国家心血管病中心 阜外医院 超声科
