铑催化剂催化烯烃不对称加氢反应研究进展
2016-03-18王红琴蒋丽红王亚明昆明理工大学化学工程学院云南昆明650500
王红琴,蒋丽红,王亚明(昆明理工大学化学工程学院,云南 昆明 650500)
铑催化剂催化烯烃不对称加氢反应研究进展
王红琴,蒋丽红,王亚明
(昆明理工大学化学工程学院,云南 昆明 650500)
摘要:不对称催化氢化反应具有完美的原子经济性和清洁高效等特点,是最受青睐的不对称合成方法之一。C=C、C=O、C=N的不对称加氢反应仍主要依赖过渡金属催化剂。过渡金属催化剂,尤其是铑催化剂,催化碳碳双键的不对称加氢反应仍是一个不断发展的领域。本文对近年来利用铑催化剂催化烯烃进行不对称氢化反应的研究进展进行了综述,着重介绍了铑-双膦配体催化体系催化烯烃不对称加氢反应的催化机理,以及铑催化剂在烯胺、不饱和羧酸及衍生物、烯醇酯和非官能团烯烃不对称氢化中的应用,并通过对现有文献的总结指出了今后铑催化剂催化烯烃氢化反应的研究重点,即:①铑-单膦配体催化烯烃不对称氢化反应的作用机理须待提出;②非官能化底物不对称催化氢化反应的手性配体亟待拓宽。
关键词:催化剂;选择性;加氢;烯烃;催化机理;配合物
第一作者:王红琴(1993—),女,硕士研究生,研究方向为工业催化和植物化工。E-mail 15198787810@163.com。联系人:蒋丽红,博士,教授,研究方向为新型催化剂在天然产物深加工中的应用。E-mail jlh65@163.com。
不对称催化加氢技术是不对称催化合成技术中发展最早、应用最成熟的手性化合物合成技术,其具有完美的原子经济性和环境友好等特点。手性化合物是合成生物活性化合物的重要中间体,已广泛用于医药工业、香料、农用化学品和精细化学品等领域。早在20世纪60年代末,孟山都公司的KNOWLES等[1]就将Wilkinson型铑膦络合物成功地用于催化烯烃不对称氢化反应,为均相不对称催化氢化反应研究奠定了基础。到了20世纪70年代中期,孟山都公司经过不断改进,最终实现了其工业化应用,生产了治疗帕金森氏症的特效药L-多巴(L-DOPA)[2]。
烯烃催化氢化为烯烃向烷烃的转化提供了最具有原子经济性的途径。烯烃根据碳碳双键上的取代基种类可以分为官能团烯烃和非官能团烯烃。官能团烯烃是指碳碳双键上带有极性基团(如氨基、酰胺基、羧基、酯基等)的一类烯烃,而非官能团烯烃指碳碳双键上的取代基仅限于脂肪族烷烃和芳香烃(如甲基、乙基、苯基等)的一类烯烃[3]。在烯烃不对称氢化反应的研究中发现,官能团烯烃往往可以得到更高的光学产率。这是因为:极性基团与催化剂的金属配位,增强了催化剂-反应物形成的配合物的刚性,从而提高了反应的对映选择性。
目前,人们致力于过渡金属配合物催化不对称氢化反应的研究[4],寻找合适的催化剂,以使不对称氢化反应在更加温和的条件下进行,并进一步扩大反应底物范围,提高反应活性和对映选择性,不少研究者也对此方面的研究进行了综述[5-6]。本文将对铑催化剂在烯烃不对称氢化领域的应用进行综述。着重介绍铑催化剂的催化机理及铑催化剂在烯胺、不饱和羧酸及其衍生物、烯醇酯和非官能团烯烃催化氢化中的应用,并对铑催化剂在不对称氢化中的发展前景作了展望。
1 铑催化烯烃不对称氢化反应机理
许多研究者对铑催化剂在不对称氢化中的作用机理进行了研究。其中,HALPERN等[7-8]对多种手性配体(如DIOP、BPPM、DIPHOS等)形成的铑催化剂在脱氢氨基酸氢化中的作用过程作了详尽的描述。HALPERN等通过反应动力学和过渡态中间体的单晶衍射,提出了铑-双膦配合物催化氢化反应机理,至今仍适用,具有一定的代表性和说服力。
HALPERN等[8]最初对非手性[Rh(DiPhos)+]催化氢化底物1(R=Me)的机理进行研究,提出了双氢机理[图1(a)]。首先,底物1和双膦配体存在快速的平衡反应;然后H2和铑发生氧化加成得到铑催化剂和底物配位中间体;随后经插入反应和还原消除反应完成整个催化氢化过程。
之后,经过更全面、更深入的研究,HALPERN等[9-10]提出了铑-手性双膦配合物催化氢化反应机理[图1(b)]。反应从离子型铑-双膦配合物3开始,形成两个催化循环,得到两种构型的产物(R构型和S构型)。该机理指出,不稳定的非对映异构加成物和H2的氧化加成反应速度比稳定的非对映异构加成物的反应速度快。很多时候可以捕获到的、量多的、稳定的过渡态中间体往往并不是反应主产物的前体,而那些量少的、不稳定的过渡态中间体,由于反应速率非常快,反而成为了主产物的真正来源。

图1 铑-双膦配体催化剂催化脱氢氨基酸不对称氢化反应机理
LANDIS等[11]通过计算模拟了Rh/Me-DuPhos催化烯酰胺不对称氢化的反应历程得到的配位模型中,MAJ和MIN对应于比较稳定和比较不稳定的Rh-底物加成物,两者自由能相差3.6kcal/mol (1kcal=4.1868kJ)。此计算结果支持了HALPERN机理中不对称氢化的主产物是由不稳定、量少的中间体(MIN)通过反锁匙模型产生的。
对于双膦配体与铑配位形成的催化剂在催化官能团烯烃氢化的作用机理,现在化学家基本都接受HALPERN推导的双氢机理。但是,对于单齿膦配体与铑配位形成的催化剂催化烯烃的反应历程,至今还没有研究者成功捕捉到过渡态中间体,反应机理尚未定论。
仔细分析非中心卡方分布的概率密度函数(Probability Density Function,PDF)可知,其在自由度大于2时总是存在一个最大值,即随机变量存在一个出现概率最大的值,这个值在所有的值中出现频次最高.因此,除了使用统计分布的期望来估计未知项以外,还可以利用这个出现概率最大的值来估计该未知项,此时的更新半径可以由下式计算得到:
2 官能团烯烃的不对称氢化反应
2.1 烯胺的不对称氢化反应
手性胺类化合物是一类重要的有机化合物,可用作拆分试剂、手性助剂、生物活性中间体等。手性过渡金属配合物催化烯胺不对称催化反应是合成手性胺及其衍生物最快捷、最简便的方法。1996年,BURK等[12]制备了双膦配体-铑络合物催化剂Rh-DuPhos、Rh-BPE,并将其用于烯胺的不对称催化氢化反应中,取得了突破性的进展。这些铑催化剂将一系列N-酰基烯胺不对称催化加氢成N-乙酰胺时具有较高的对映选择性(高达97.8%ee)。随后,这些铑催化剂也被用于α-烷基烯胺和环烯胺的不对称加氢反应中,其在α-烷基烯胺的不对称氢化反应中获得了较高的催化活性和对映选择性[13]。
自BURK将铑络合物催化剂Rh-DuPhos、Rh-BPE用于烯胺的不对称催化氢化反应后,研究者开发了大量的1,2-二膦配体,并成功地用于不对称加氢反应中,如手性胺、手性醇的合成。JIA等[14]和VAN DEN BERG等[15]曾报道过MonoPhos配体的铑配合物在N-酰基烯胺的不对称加氢反应中具有优异的对映选择性。在2MPa H2、20℃、CH2Cl2为溶剂的条件下,采用催化剂Rh-MonoPhos ([Rh(COD)2]BF4∶MonoPhos =1∶2)催化底物N-酰基烯胺不对称加氢,其产物N-乙酰基胺具有70%~96%的ee值。在配体MonoPhos的3-和3'-位引入两个甲基基团增大了配体的空间位阻,以至于对映选择性急剧下降,用此配体来催化N-酰基烯胺的不对称加氢时,仅有44%的对映选择性,且产物构型与MonoPhos配体作催化剂时所获得的产物构型相反。反之,在配体MonoPhos的6-和6'-位引入取代基(如Br)时对对映选择性影响不大[16]。此外,JIA等[17]发现:当用二乙氨基来取代MonoPhos配体上的二甲氨基时,能够明显提高N-酰基烯胺不对称加氢产物的对映选择性(99%ee)。BERNSMANN等[18]对MonoPhos配体上二烷基氨基基团进行了系统的研究,结果表明,对于联萘基型手性单磷酰胺配体来说,哌啶(PipPhos)和吗啉(MorfPhos)是最适宜的二烷基氨基基团,在室温下它们能将反应的对映选择性提高至99%。总之,手性单磷酰胺配体中的刚性骨架有利于烯酰胺的不对称加氢反应。LI等[19]用配体(RC,RFc)-Walphos来催化烯胺的不对称加氢反应,获得了96.2%的对映选择性,比MonoPhos配体所获得的对映选择性高。他们也对八氢萘结构的配体进行了研究,结果表明,磷原子上含有哌啶基(98%ee)和吗啉基(97%ee)的配体在烯胺不对称加氢反应中所获得的对映选择性较高。
2012年,段正超等[20]报道了衍生于手性双二茂铁乙胺的手性单齿亚磷酰胺配体的铑络合物催化剂在N-酰基烯胺的不对称氢化反应中的应用。研究发现,在N-酰基烯胺的不对称催化氢化反应中,(R,R,S)-单齿亚磷酰胺配体形成的催化剂表现出了较好的催化活性及中等的手性诱导能力。配体的中心手性与轴手性是否匹配对氢化产物的对映选择性影响较大,其中手性(R,R,S)-单齿亚磷酰胺配体获得了59%的对映选择性;而非手性(R,R,R)-单齿亚磷酰胺配体只获得了14%的对映选择性。随后,作者以(R,R,S)-单齿亚磷酰胺为配体,考察了其在β-取代的N-酰基烯胺的不对称氢化反应中的情况,发现(R,R,S)-单齿亚磷酰胺配体对一系列烯胺底物均具有较好的手性诱导能力,获得了90%以上的对映选择性,底物中苯环上取代基的电子效应对反应活性及对映选择性均没有明显的影响。
2013年,LI等[21]大规模地合成了P-手性膦配体MeO-BIBOP,并且原位修饰制备了其铑配合物[Rh(nbd)(MeO- BIBOP)]BF4,此外,将其用于N-乙酰基烯胺的不对称催化氢化反应中,考察了P-手性膦配体MeO-BIBOP的催化性能。在不对称氢化反应过程中,由于考虑到底物的溶解性,选取甲醇作为溶剂。结果表明,反应温度、反应压力对反应活性和对映选择性没有太大的影响。在优化条件下,甚至当催化剂的负载量降低至0.0005%时,对映选择性仍几乎保持不变,证明了该催化系统具有显著的鲁棒性。作者对加氢后的产品进行结晶、分离,其分离产率为84%,产品的对映体比率为99.9∶0.1。此外,作者认为该方法能够通用于医药、化学品中各种重要手性胺的合成。
2014年,JIANG等[22]合成了膦配体TangPhos及其铑络合物,并应用于β-乙酰氨基丙烯腈砜的不对称催化氢化反应中(图2)。结果表明,Z构型β-乙酰氨基丙烯腈砜转化率和对映选择性比E构型的高。此外,该作者还对反应条件进行了优化,适宜反应条件为:S∶C=100∶1(底物∶催化剂=100∶ 1),室温,5MPa H2,甲醇为溶剂,反应时间为12h。在优化条件下,对具有不同取代基的Z构型底物进行了研究,结果表明,当R1为芳基,R2为烷基取代基时,都获得了良好的转化率和对映选择性(84%~94% ee);当R1、R2均为芳基取代基时,对映选择性有所提高(97% ee),这可能是因为芳基的空间位阻有利于对映选择性的提高;而当R1、R2都为烷基时,对映选择性比较低,只有23%。

图2 TangPhos配体及β-乙酰氨基丙烯腈砜的不对称催化氢化反应
2.2 不饱和羧酸及其衍生物的不对称氢化反应
不饱和羧酸及其衍生物主要包括不饱和羧酸、不饱和酯等,该类化合物的不对称氢化反应能够生成一系列有价值的手性羧酸衍生物,在医药、农药、香料等精细化学品的合成中具有广泛的应用[23-24]。因此,α,β-不饱和羧酸及其衍生物的不对称氢化反应受到了广泛地关注。
不同研究团队开展了对铑催化α-异丙基肉桂酸不对称氢化反应的研究,该反应能够获得合成手性药物肾素抑制剂阿利吉仑的重要中间体[25]。研究发现,以3,3'-二甲基-PipPhos为配体,在55℃、8MPa H2条件下,反应过程中加入三苯基膦时,由于三苯基膦的协同效应,能显著地改善反应速率和选择性,在优化条件下,原位修饰的铑催化体系催化α-异丙基肉桂酸加氢反应能获得97%收率及90%ee,该过程已实现大规模工业化生产[26-27]。在此之前,WEISSENSTEINER等[28]在温和的条件下,使用配体(RC,RFc)-Walphos合成的铑催化剂催化α-异丙基肉桂酸加氢也获得类似的催化效率。CHEN等[29]使用配体(RC,RC,SFc,SFc,SP,SP)-TriFer原位合成的铑基催化剂催化α-异丙基肉桂酸加氢反应时,产物的对映选择性高于98%,作者推测获得如此高的对映选择性可能是由于配体的二甲基氨基与底物的羧基间具有静电作用。此外,ANDRUSHKO等[30]筛选了一些市售的光学纯二茂铁双膦配体并用于α-异丙基肉桂酸的加氢反应中。采用铑基前体时,该反应能获得95%的对映选择性,而采用钌基前体时仅有42%的对映选择性。
2008年,YANG等[31]合成了新型的(R)-BINOL衍生的聚合物单齿亚磷酰胺配体,并将其用于铑催化的脱氢氨基酸衍生物不对称加氢反应中,获得了较高的对映选择性(99%ee)和转化率。研究发现,由于其独特的结构,含有刚性高分子链的聚合物配体能够阻止高分子中Rh和亚磷酰胺基团之间的配位。随着配体含量的增加,将会形成沉淀并导致转化率和对映选择性降低。
2011年,FARKAS等[32]以羟烷基磷和BINOL衍生的氯膦酸为原料,制备了一种新型的联萘酚基膦-亚磷酸酯配体,并通过原位修饰制备了阳离子铑配合物,将其用于脱氢氨基酸酯和衣康酸二甲酯的不对称催化氢化反应中。研究表明,底物对选择性和反应速率有着显著的影响。配体在衣康酸二甲酯的不对称催化氢化反应中具有良好的对映选择性,高达99.4%。刘龑等[33]研究了手性单齿亚磷酰胺配体DpenPhos在Rh(I)催化的E-和Z-构型β-脱氢氨基酸酯不对称氢化中的应用。结果表明,N原子上含有H的亚磷酰胺配体与Rh(I)形成的催化剂通常比N原子上不含H的配体表现出更高的反应活性。在E-构型β-脱氢氨基酸酯的不对称氢化反应中,催化剂可以实现底物的常压催化氢化,取得了92%~96%的对映选择性,催化剂用量可降低至0.2%;对于Z-构型β-脱氢氨基酸酯的不对称氢化反应,尤其是β-芳基取代衍生物的氢化反应,氢化产物的ee值可以达到96%~98%。该类催化剂为天然或非天然光学活性β-氨基酸的合成提供了一种简便、高效的方法。
2012年,IMAMOTO Tsuneo等[34]以(S)-和(R)-叔丁基甲基膦-硼烷作为关键中间体合成了P-手性二膦配体QuinoxP*、BenzP*和DioxyBenzP*,这些配体具有较高的刚性。该作者将其配合物用来催化α-/β-脱氢氨基酸衍生物的不对称加氢反应。在甲醇为溶剂、S/C=10000、0.3MPa H2、室温、0.3~0.5h的反应条件下催化α-乙酰氨基肉桂酸甲酯不对称加氢,以QuinoxP*-Rh为催化剂时,底物的转化率较低,但产物的ee值较高,达到99.7%;而以BenzP*-Rh 和DioxyBenzP*-Rh为催化剂时,底物获得了较高的转化率,但产物的ee值较低。在β-脱氢氨基酸衍生物的不对称催化加氢中,大多数情况下,E构型衍生物不对称加氢产物的对映选择性比Z构型的高。此外,这3种配体可用于几种药物中间体的合成中,如δ-阿片受体拮抗剂Dmt-tic、VLA-4拮抗剂S9059、GAVA-B拮抗剂CGP-55845等,证明了它们在不对称加氢反应中的实用性。
GHEORGHIU等[35]通过吡咯烷基双膦配体的共价键将铑配合物[Rh(COD)2]BF4固载到官能化的碳纳米管上,制备了负载型手性铑催化剂,并将其用于2-乙酰氨基丙烯酸甲酯和α-乙酰氨基肉桂酸的不对称催化氢化反应中。研究表明,在2-乙酰氨基丙烯酸甲酯的不对称氢化反应中,该催化剂具有一定的对映选择性,且可重复利用;对于α-乙酰氨基肉桂酸的不对称氢化反应,该催化剂具有良好的催化活性,且高于均相催化剂[Rh(COD)2]BF4的催化活性。
2014年,苗晓等[36]合成了基于甘露醇骨架的新型tropos配体,以及甘露醇和1,1'-联-2-萘酚为原料制备的双齿亚磷酸酯配体,将这些配体应用于铑催化α-脱氢氨基酸酯衍生物的不对称氢化中。在0.0025mmol[Rh(cod)2]BF4、配体/Rh=1.1、底物/Rh= 100、3MPa H2、2mL CH2Cl2、室温的条件下反应15h,含有双亚异丙基甘露醇骨架的配体对α-乙酰氨基肉桂酸甲酯氢化反应的对映选择性达85.6%,转化率大于99.9%。研究表明,具有亚异丙基甘露醇骨架的配体显示出更好的立体诱导能力;配体的联萘酚构型决定产物的构型,而tropos配体的甘露醇骨架决定氢化产物的构型。此外,该作者还对底物进行了拓宽,结果表明,当供电子基团在芳环的邻位和对位时,其氢化产物的对映选择性均高于相应位置为吸电子基团的,这与HUANG等[37]报道的结果相一致。
2.3 烯醇酯的不对称氢化反应
烯醇酯的不对称氢化反应为合成手性酯提供了一种简便的方法,有望取代前手性酮不对称还原合成手性酯。LIU等[38]采用原位修饰的方法合成了Rh (I)/DpenPhos催化剂,并用于α-芳基和α-烷基烯醇酯的不对称氢化反应。以1,2-二氯乙烷为溶剂,在n(DpenPhos)/n(Rh)=2、0.5MPa H2、室温的条件下反应10h,烯醇酯几乎全部转化,产物对映选择性高达87%~95%。2013年,KLEMAN等[39]采用膦-亚磷酸盐(P-OP)配体修饰铑前体,形成了一系列的铑催化剂([Rh(NBD)(P-OP)]BF4)(NBD= Norbornadiene,降莰二烯),并将其用于烯醇酯的加氢反应中(图3)。以CH2Cl2为溶剂,在2MPa H2、n(底物)/n(催化剂)=100、室温的条件下反应24h,底物a几乎全部转化,且获得了较高的对映选择性(88%~89%ee)。该作者发现,在相同条件下,具有对甲苯结构的催化剂的手性诱导能力高于具有异丙醇结构催化剂,且稍微提高温度对其产物的对映选择性影响不大。
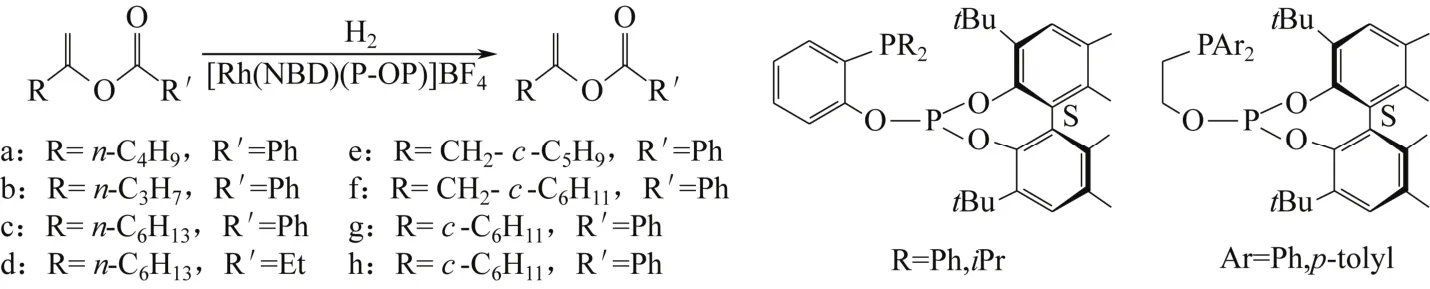
图3 膦-亚磷酸盐(P-OP)配体及烯醇酯不对称加氢反应
单齿亚磷酰胺配体是一类高效的单齿磷配体,尤其是PipPhos配体和八氢萘单磷酰胺配体。PANELLA等[40]成功地将联萘酚(BINOL)衍生的手性单齿亚磷酰胺配体PipPhos应用在Rh-催化芳香烯醇乙酸酯和芳香烯醇氨基甲酸酯的不对称氢化反应中,考察了溶剂种类对反应结果的影响,结果表明,CH2Cl2是最适合该反应体系的溶剂。以MeOH为溶剂时,产物的对映选择性较高,但转化率低,且产物构型与CH2Cl2为溶剂时的产物构型相反。在0.5MPa H2、0.2mmol底物、4mLCH2Cl2、n(配体)/n([Rh(COD)2]BF4)=2,室温的条件下反应16h,两种烯醇酯几乎全部转化,产物对映选择性高达96%~98%。2007年,WANG等[41]利用一类基于1,2,3,4-四氢-1-萘胺骨架的手性膦-亚磷酰胺酯配体(Rc,Ra)-THNA-Phos,成功实现了芳基取代的α-烯醇酯膦酸酯的Rh-催化不对称氢化,产物的对映选择性高达99%,但该配体优化困难,难以在N原子和苯环上引入其他基团;且与铑形成的催化剂催化活性不高,在芳基取代的α-烯醇酯膦酸酯的不对称氢化中,底物与催化剂比值最高只能达到1000。2008年,YU等[42]以1-氨基萘为原料,成功合成了新型手性膦-亚磷酰胺配体(S)-HY-Phos,并用于铑催化α-烯醇膦酸酯不对称氢化反应中。以CH2Cl2为溶剂,在0.5mmol底物、n(底物)/n(催化剂)=100、配体/Rh=1.1(摩尔比)、2MPa H2、室温的条件下反应24h,底物几乎全部转化,对映选择性达98%以上。该配体不含手性中心,但对于铑催化的α-烯醇膦酸酯不对称氢化反应,其催化性能比以1,2,3,4-四氢-1-氨基萘为原料制备的、具有手性中心的配体(R,R)-THNAPhos的催化性能要好。
2012年,胡娟等[43]还合成了苯乙胺衍生的手性膦-亚磷酰胺酯配体(Sc,Sa)-PEAPhos,并将其用于Rh-催化α-烯醇酯膦酸酯的不对称氢化反应中,考察了配体结构及反应条件对反应结果的影响,研究表明,配体结构对Rh-催化α-烯醇酯膦酸酯不对称氢化反应具有很大的影响,该类配体中(S)-中心手性和(S)-轴手性是匹配的构型;当在配体氮原子上引入甲基后,配体的立体选择性得到显著提高。此外,该作者在优化条件下研究了各种底物的适用范围,结果表明,该类膦-亚磷酰胺酯配体对烷基取代和苯基取代的α-烯醇酯膦酸酯底物显示出优异的立体选择性,对映选择性都达到或超过99%。与配体(Rc,Ra)-THNA-Phos相比,配体(Sc,Sa)-PEAPhos显示出类似的对映选择性及更高的催化活性,且合成成本更低。
3 非官能化烯烃的不对称氢化反应
前面讨论的烯烃不对称氢化反应中,获得高对映选择性的反应底物都是带有官能团(酰胺基、羧基、酯基、羟基等)的烯烃。很长一段时间里,利用铑催化剂催化的非官能化烯烃的不对称氢化反应均未能获得理想的对映选择性。因此,非官能化烯烃不对称氢化反应的发展远远滞后于官能化烯烃不对称氢化反应的发展。之后,非官能化烯烃的不对称加氢也逐渐地发展起来,但所用的催化剂多为铱催化剂。
SUAREZ等[44]使用Rh(PPh3)3Cl和[Rh(COD)2] BF4(COD=1,5-环辛二烯)作为催化剂,以[BMIM]BF4离子液体为反应介质,催化环己烯进行加氢反应,分析结果发现,离子型[Rh(COD)2]BF4为催化剂能使环己烯全部转化为环己烷,而Rh(PPh3)3Cl为催化剂时仅获得40%的转化率。2001年,LIU等[45]使用离子液体和超临界二氧化碳为介质,以Rh(PPh3)3Cl为催化剂催化1-癸烯加氢反应,研究发现,在反应温度50℃、氢气压力为4.8MPa下,反应1h,其转化率为98%。
PEREZ-CADENAS等[46]通过共价键作用将Rh(PPh3)3Cl固载在活性炭和多壁碳纳米管上,制备了负载型催化剂,并用于环己烯的不对称加氢反应中。结果表明,多壁碳纳米管负载型催化剂的催化活性明显高于均相Rh(PPh3)3Cl催化剂的催化活性,活性炭负载型催化剂的催化活性与均相Rh(PPh3)3Cl催化剂的催化活性相近,但是活性炭负载型催化剂比均相Rh(PPh3)3Cl催化剂易回收分离。
SILBERNAGEL等[47]首先采用(EtO)3Si(CH2)3PPh2对ZrP进行了改性,然后再通过配体的交换作用将Rh(PPh3)Cl固载在改性后的ZrP表面,形成了负载型催化剂,其中配体和催化剂并没有插层在ZrP载体中,而是通过膦基团链接在载体上的。该作者将其用于1-十二烯的加氢反应中,该催化剂具有较高的催化活性,反应后以间歇方式回收,可循环使用15次。此外,该催化体系中的交联剂(EtO)3Si(CH2)3PPh2还可以用异氰酸酯、环氧化合物、磷酸盐和膦酸等来代替。
本文作者课题组[48]采用可溶性铑盐和过量的三苯基膦乙醇溶液加热回流的方法制备了Rh(PPh3)3Cl催化剂,研究了咪唑类离子液体-Rh(PPh3)3Cl催化体系催化α-蒎烯不对称加氢反应。在催化剂用量5%、反应温度120℃、反应压力1.5MPa、反应时间3h条件下,α-蒎烯的转化率为99.29%,顺式蒎烷的对映选择性为98.42%。
4 结语与展望
近年来,由于人们对手性物质需求的增加,不对称加氢成为人们研究的热门课题之一。综上所述,铑催化不对称加氢反应作为一种最经济、最清洁、最高效的不对称合成方法已经取得了很大的进展。在这方面的研究中,可通过改变配体的类型及取代基的空间结构来提高催化剂的催化活性,以适应不同的催化氢化反应的需求,为合成不同类型的手性化合物提供有效的方法。
目前,在不对称催化氢化领域,虽然已经有很多手性配体和催化剂报道,但整体而言,仍有很多不对称氢化反应不能实现高效的手性诱导;另外,还有许多潜在的不对称催化氢化反应仍待发现和建立。因此,发展新颖高效手性配体及催化剂,以及高效不对称催化氢化新反应仍是不对称催化氢化领域今后的研究重点;Rh-单膦配体催化烯烃不对称氢化反应的作用机理须待提出。此外,非官能化底物不对称催化氢化反应的手性配体及前体亟待拓宽。不对称催化氢化反应研究将会为手性化合物提供更多高效、高选择性、原子经济、环境友好的不对称合成方法。
参 考 文 献
[1] KNOWLES W S,SABACKY M J. Catalytic asymmetric hydrogenation employing a soluble,optically active,rhodium complex[J]. Chemical Communications,1968,22:1445-1446.
[2] KNOWLES W S. Application of organometallic catalysis to the commercial production of L-DOPA[J]. Journal of Chemical Education,1986,63(3):222-225.
[3] 陈传杰,魏作君. 铱-氮膦配体催化剂在非官能化烯烃不对称氢化中的研究[J]. 化学进展,2009,21(5):990-996.
[4] INAGAKI Fuyuhiko,MUKAI Chisato. Rhodium(I)-catalyzed intramolecular pauson-khand-type [2+2+1] cycloaddition of allenenes[J]. Org. Lett.,2006,8(6):1217-1220.
[5] 吴跃,薛屏. 多相手性催化剂的制备及其对不对称加氢反应的催化作用[J]. 化工进展,2006,25(11):1301-1308.
[6] 何年志,张学勤,肖美添. 手性修饰催化剂在多相不对称氢化中的研究进展[J]. 化工进展,2012,31(12):2694-2701,2719.
[7] HALPERN J. Mechanism and stereoseIectivity of asymmetric hydrogenation[J]. Science,1982,217(4558):401-407.
[8] CHAN A S C,HALPERN J. Interception and characterization of a hydridoalkylrhodium intermediate in a homogeneous catalytic hydrogenation reaction[J]. Journal of the American Chemical Society,1980,102(2):838-840.
[9] LANDIS C R,HALPERN J. Asymmetric hydrogenation of methyl (Z)-alpha-acetamidocinnamate catalyzed by [1,2-bis(phenyl-o-anisoyl) phosphino) ethane] rhodium (I):kinetics,mechanism and origin of enantioselection[J]. Journal of the American Chemical Society,1987,109(6):1746-1754.
[10] CHAN A S C,PLUTH J J,HALPERN J. Identification of the enantioselective step in the asymmetric catalytic hydrogenation of a prochiral olefin[J]. Journal of the American Chemical Society,1980,102(18):5952-5954.
[11] LANDIS C R,FELDGUS S. A simple model for the origin of enantioselection and the anti “Lock-and Key” motif in asymmetric hydorgenation of enamides as catalyzed by chiral diphosphine complexes of Rh(I)[J]. Angew. Chem. Int. Ed.,2000,39(16):2863-2866.
[12] BURK Mark J,WANG Yanming,LEE Jeffrey R. A convenient asymmetric synthesis of α-1-arylalkylamines through the enantioselective hydrogenation of enamides[J]. J. Am. Chem. Soc., 1996,118(21):5142-5143.
[13] BURK MARK J,CASY Guy,JOHNSON Nicholas B. A three-step procedure for asymmetric catalytic reductive amidation of ketones[J]. J. Org. Chem.,1998,63(18):6084-6085.
[14] JIA Xian,GUO Rongwei,LI Xingshu,et al. Highly enantioselective hydrogenation of enamides catalyzed by rhodium-monodentate phosphoramidite complex[J]. Tetrahedron Letters,2002,43(32):5541-5544.
[15] VAN DEN BERG M,HAAK Robert M,MINNAARD Adriaan J,et al. Rhodium/monophos-catalyzed asymmetric hydrogenation of enamides[J]. Adv. Synth. Catal.,2002,344(9):1003-1007.
[16] VAN DEN BERG Michel,HAAK Robert M,MINNAARD Adriaan J,et al. Monodentate phosphoramidites:a breakthrough in rhodium-catalysed asymmetric hydrogenation of olefins[J]. Adv. Synth. Catal.,2003,345(1):308-323.
[17] JIA Xian,LI Xingshu,XU Lijin,et al. Highly efficient rhodium/monodentate phosphoramidite catalyst and its application in the enantioselective hydrogenation of enamides and α-dehydroamino acid derivatives[J]. J. Org. Chem.,2003,68(11):4539-4541.
[18] BERNSMANN Heiko,VAN DEN BERG Michel,HOEN Rob,et al. Pipphos and morfphos:privileged monodentate phosphoramidite ligands for rhodium-catalyzed asymmetric hydrogenation[J]. J. Org. Chem.,2005,70(3):943-951.
[19] LI Xingshu,JIA Xian,LU Gui,et al. Highly enantioselective hydrogenation of enamides catalyzed by rhodium-monodentate phosphoramidite complex derived from H8-BINOL[J]. Tetrahedron:Asymmetry,2003,14(18):2687-2691.
[20] 段正超,王联芝,胡向平,等. 衍生于手性双二茂铁乙胺的单齿亚磷酰胺配体在不对称催化氢化中的应用[J]. 分子催化,2012,26(4):328-332.
[21] LI Wenjie,RODRIGUEZ Sonia,DURAN Adil,et al. The P-chiral phosphane ligand (MeO-BIBOP) for efficient and practical large-scale Rh-catalyzed asymmetric hydrogenation of N-acetyl enamides with high TONs[J]. Org. Process Res. Dev.,2013,17(8):1061-1065.
[22] JIANG Jun,WANG Yan,ZHANG Xumu. Rhodium-catalyzed asymmetric hydrogenation of β-acetylamino acrylosulfones:a practical approach to chiral β-amido sulfones[J].ACS Catal.,2014,4 (5):1570- 1573.
[23] BLASER Hans-Ulrich,PUGIN Benoît,SPINDLER Felix. Progress in enantioselective catalysis assessed from an industrial point of view[J]. Journal of Molecular Catalysis A:Chemical,2005,231(1-2):1-20.
[24] VITTORIO Farina,REEVES Jonathan T,SENANAYAKE Chris H,et al. Asymmetric synthesis of active pharmaceutical ingredients[J]. Chem. Rev.,2006,106(7):2734-2793.
[25] FRAMPTON James E,CURRAN Monique P. Aliskiren:a review of its use in the management of hypertension[J]. Drugs,2007,67(12):1767-1792.
[26] LEFORT Laurent,BOOGERS Jeroen A F,DEVRIES André H M,et al. High throughput screening of monophos instant ligand library leads to a ton-scale asymmetric hydrogenation process[J]. Topics in Catalysis,2006,40(1/2/3/4):185-191.
[27] BOOGERS Jeroen A F,FELFER Ulfried,KOTTHAUS Martina,et al. A mixed-ligand approach enables the asymmetric hydrogenation of an α-isopropylcinnamic acid en route to the renin inhibitor aliskiren[J]. Org. Process Res. Dev.,2007,11(3):585-591.
[28] STURM Thomas,WEISSENSTEINER Walter,SPINDLER Felix,et al. A novel class of ferrocenyl-aryl-based diphosphine ligands for Rh- and Ru-catalysed enantioselective hydrogenation[J]. Adv. Synth. Catal.,2003,345(1/2):160-164.
[29] CHEN Weiping,MCCORMACK Peter J,MOHAMMED Karim,et al. Stereoselective synthesis of ferrocene-based C2-symmetric diphosphine ligands:application to the highly enantioselective hydrogenation of alpha-substituted cinnamic acids[J]. Angew. Chem.,2007,119(22):4219-4222.
[30] ANDRUSHKO Natalia,ANDRUSHKO Vasyl,THYRANN Thomas,et al. Synthesis of enantiopure (R)-2-[4-methoxy-3-(3-methoxypropoxy)-benzyl]-3-methylbutanoic acid—a key intermediate for the preparation of Aliskiren[J]. Tetrahedron Letters,2008,49(41):5980-5982.
[31] YANG Meng,ZHANG Ji,WANG Na,et al. A new class of polymeric monodentate phosphoramidite ligands for asymmetric hydrogenation of α-dehydroamino acid derivatives[J]. ARKIVOC,2008,28(16):279-287.
[32] FARKAS Gergely,BALOGH Szabolcs,SZÖLLŐSYET Áron,et al. Novel phosphine-phosphites and their use in asymmetric hydrogenation[J]. Tetrahedron:Asymmetry,2011,22(24):2104-2109.
[33] 刘龑,王正,丁奎岭. DpenPhos/Rh(I)催化的β-脱氢氨基酸酯的不对称氢化反应[J]. 化学学报,2012,70(13):1464-1470.
[34] IMAMOTO Tsuneo,TAMURA Ken,ZHANG Zhenfeng,et al. Rigid P-chiral phosphine ligands with tert-butylmethylphosphino groups for rhodium-catalyzed asymmetric hydrogenation of functionalized alkenes[J]. J. Am. Chem. Soc.,2012,134(3):1754-1769.
[35] GHEORGHIU C C,DE LECEA C S M,MACHADO B F,et al. Chiral rhodium complexes covalently anchored on carbon nanotubes for enantioselective hydrogenation[J]. Dalton Trans.,2014,43(20):7455-7463.
[36] 苗晓,李海峰,逄增波,等. D-甘露醇衍生的手性亚磷酸酯配体:合成及应用于铑催化α-脱氢氨基酸酯不对称氢化[J]. 分子催化,2014,28(5):393-399.
[37] HUANG H M,ZHENG Z,LUO H L,et al. A novel class of P-O monophosphite ligands derived from D-mannitol:broad applications in highly enantioselective Rh-catalyzed hydrogenations[J]. The Journal of organic chemistry,2004,69(7):2355-2361.
[38] LIU Y,WANG Z,DING K. Rh (I)/DpenPhos catalyzed asymmetric hydrogenation of enol esters and potassium (E)-3-cyano-5-methylhex-3- enoate[J]. Tetrahedron,2012,68(37):7581-7585.
[39] KLEMAN P,GONZÁLEZ-LISTE P J,GARCÍA-GARRIDO S E,et al. Highly enantioselective hydrogenation of 1-alkylvinyl benzoates:a simple,nonenzymatic access to chiral 2-alkanols[J]. Chemistry—A European Journal,2013,19(48):16209-16212.
[40] PANELLA L,FERINGA B L,DE VRIES J G,et al. Enantioselective Rh-catalyzed hydrogenation of enol acetates and enol carbamates with monodentate phosphoramidites[J]. Organic Letters,2005,7 (19):4177-4180.
[41] WANG D Y,HU X P,HUANG J D,et al. Highly enantioselective synthesis of α-hydroxy phosphonic acid derivatives by Rh-catalyzed asymmetric hydrogenation with phosphine-phosphoramidite ligands[J]. Angewandte Chemie,2007,119(41):7956-7959.
[42] YU S B,HUANG J D,WANG D Y,et al. Novel chiral phosphine-phosphoramidite ligands derived from 1-naphthylamine for highly efficient Rh-catalyzed asymmetric hydrogenation[J]. Tetrahedron:Asymmetry,2008,19(15):1862-1866.
[43] 胡娟,王道永,郑卓,等. 苯乙胺衍生的手性膦-亚磷酰胺酯配体在 Rh-催化α-烯醇酯膦酸酯的不对称氢化反应中的应用[J]. 分子催化,2012,26(6):487-492.
[44] SUAREZ P A Z,DULLIUS J E L,EINLOFT S,et al. The use of new ionic liquids in two-phase catalytic hydrogenation reaction by rhodium complexes[J]. Polyhedron,1996,15(7):1217-1219.
[45] LIU F,ABRAMS M B,BAKER R T,et al. Phase-separable catalysis using room temperature ionic liquids and super critical carbon dioxide[J]. Chemical Communications,2001,1(5):433-434.
[46] PEREZ-CADENAS M,LEMUS-YEGRES L J,ROMÁNMARTÍNEZ M C,et al. Immobilization of a Rh complex derived from the Wilkinson’s catalyst on activated carbon and carbon nanotubes[J]. Applied Catalysis A:General,2011,402(1-2):132-138.
[47] SILBERNAGEL Rita,DÍAZ Agustín,STEFFENSMEIERET Eric,et al. Wilkinson-type hydrogenation catalysts immobilized on zirconium phosphate nanoplatelets[J]. Journal of Molecular Catalysis A:Chemical,2014,394:217-223.
[48] 蒋丽红. 一种由α-蒎烯不对称催化加氢制备顺式蒎烷的方法:201410183712.1[P]. 2014-05-05.
综述与专论
Research progress of olefins asymmetric hydrogenation catalyzed by rhodium catalysts
WANG Hongqin,JIANG Lihong,WANG Yaming
(Faculty of Chemical Engineering,Kunming University of Science and Technology,Kunming 650500,Yunnan,China)
Abstract:Asymmetric hydrogenation has the advantage of cleanliness,perfect atom economy,and is one of the hottest methods of asymmetric synthesis. The asymmetric hydrogenation in C=C,C=O,C=N are still primarily dependent on the use of transition metal catalysts. The study of transition metal catalysts,especially the asymmetric hydrogenation of carbon-carbon double bond catalyzed by rhodium catalysts is still an evolving field. In the present review,the progress on asymmetric hydrogenation of olefins catalyzed by rhodium catalysts are described,including the catalytic mechanism of rhodium-diphosphine ligand catalyst system,the application of asymmetric hydrogenation of enamines,unsaturated carboxylic acids and their derivatives,enol ester as well as unfunctionalized olefins catalyzed by rhodium catalysts. The development trend of rhodium catalysts for asymmetric hydrogenation of olefins was pointed out. For instance:① the catalytic mechanism of asymmetric hydrogenation of olefins by rhodium-monophosphine ligand needs to be understood; ②more chiral ligands for asymmetric hydrogenation of unfunctionalized olefins are still wanted.
Key words:catalyst; selectivity; hydrogenation; olefin; catalytic mechanisms; complexes
基金项目:国家自然科学基金项目(21266012)。
收稿日期:2015-05-07;修改稿日期:2015-07-09。
DOI:10.16085/j.issn.1000-6613.2016.02.021
中图分类号:O 643
文献标志码:A
文章编号:1000–6613(2016)02–0485–08
