HPLC法测定萘普生固体脂质纳米粒中主成分的含量Δ
2016-03-11高赛男周学刚孙世芹哈尔滨医科大学大庆校区药学院黑龙江大庆163319
付 佳,高赛男,周学刚,辛 萍,孙世芹(哈尔滨医科大学大庆校区药学院,黑龙江大庆 163319)
HPLC法测定萘普生固体脂质纳米粒中主成分的含量Δ
付 佳*,高赛男,周学刚,辛 萍,孙世芹#(哈尔滨医科大学大庆校区药学院,黑龙江大庆 163319)
目的:建立测定萘普生固体脂质纳米粒中主成分含量的方法。方法:采用微乳法制备萘普生固体脂质纳米粒,并采用高效液相色谱法测定含量。色谱柱为Elite C18,流动相A为甲醇、流动相B为0.01 mol/L磷酸二氢钾缓冲溶液(磷酸调节pH至3.0)(梯度洗脱),流速为1.0 ml/min,检测波长为240 nm,柱温为25Ⅱ,进样量为20µl。结果:萘普生检测质量浓度线性范围为0.2~180.0µg/ml(r=0.999 7);精密度、稳定性、重复性试验的RSD<1%;加样回收率为98.97%~100.67%,RSD=0.50%(n=9)。结论:该方法简便、精确,可排除萘普生固体脂质纳米粒中辅料的干扰,适用于其中主成分的含量测定。
萘普生固体脂质纳米粒;高效液相色谱法;含量测定
萘普生(Naproxen,Nap)又名甲氧异丙酸,化学名(+)-α-甲基-6-甲氧基-2-萘乙酸,是一种非甾体抗炎药,具有明显的抑制前列腺素合成的作用,可使前列腺素的释放减少甚至停止,因而有较强的消炎、解热和镇痛作用,临床主要用于治疗风湿性关节炎、类风湿性关节炎及强直性脊椎炎等[1]。Nap的常用剂型有片剂、胶囊、颗粒剂和注射剂等,而使用这些剂型可能出现不同程度的胃部不适、胃溃疡及心血管系统不良反应[2]。固体脂质纳米粒(Solid lipid nanoparticles,SLN)是一种新型的纳米粒给药系统,具有物理稳定性高、可控制药物释放、可避免药物降解或泄漏以及靶向性好、无毒等优点[3-4]。笔者以硬脂酸为油相,大豆磷脂为表面活性剂,牛黄胆酸钠为助表面活性剂[5],利用微乳法[6]成功制备了Nap固体脂质纳米粒(Nap-SLN),可避免使用Nap常规口服或注射剂型可能引发的不良反应,从而利于达到更好的治疗效果。此前国内外已有采用紫外分光光度法(UV)和高效液相色谱法(HPLC)测定Nap含量的文献报道[7-8]。但是,由于Nap-SLN中辅料的存在,沿用药典或文献报道的方法测定其中主成分的含量可能会存在干扰。因此,本试验在已有的国内外相关文献报道基础上探索了新的测定Nap-SLN中主成分含量的HPLC法,旨在为后续研究奠定基础。
1 材料
1.1 仪器
LC-20AT型HPLC仪,包括SIL-20A型自动进样器、LC solution色谱工作站等(日本岛津公司);KH-500SPV型超声波清洗器(昆山禾创超声仪器有限公司);DF-101S型集热式恒温加热磁力搅拌器(河南省予华仪器有限公司);HH-1型数显恒温水浴锅(金坛市荣华仪器制造有限公司);JY92-11型超声波细胞粉碎机(宁波新芝生物科技股份有限公司);H1650-W型
ठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठठ台式微量高速离心机(长沙湘仪离心机仪器有限公司);SHZ-D(Ⅱ)型循环水式真空泵(河南省予华仪器有限公司)。
1.2 药品与试剂
Nap对照品(中国食品药品检定研究院,批号:100198-201205,纯度:99.6%);Nap原料药(西安昊轩生物科技有限公司,批号:10061212,纯度:98%);Nap-SLN(哈尔滨医科大学大庆校区药学院自制,批号:20150801、20150802、20150803);甲醇为色谱纯,其他试剂均为分析纯,水为超纯水。
2 方法与结果
2.1 Nap-SLN的制备
在查阅大量文献的基础上[9],拟定SLN的处方为:硬脂酸199 mg、大豆磷脂108 mg、牛黄胆酸钠355 mg。应用微乳法制备Nap-SLN,以纳米粒的粒径分布为考察指标分别对超声分散功率、超声分散时间、磁力搅拌温度、磁力搅拌强度、磁力搅拌时间及稀释倍数等因素进行单因素试验,由此确定制备Nap-SLN的最佳工艺条件具体为:磁力搅拌温度控制在70Ⅱ,将硬脂酸和Nap原料药混合熔融作为油相,在600 r/min磁力搅拌速度下,将相同温度的含有表面活性剂的水相大豆磷脂和牛黄胆酸钠分别滴入油相中,滴加完毕后,继续搅拌20 min,形成初乳。将初乳加至1 ml的2~3Ⅱ冷水中,超声(功率:200 W,频率:25 kHz)分散3 min,得Nap-SLN分散液,冷冻干燥24 h,即得Nap-SLN冻干粉。
2.2 色谱条件
色谱柱:Elite C18(250 mm×4.6 mm,5µm);流动相A为甲醇、流动相B为0.01mol/L磷酸二氢钾缓冲溶液(磷酸调节pH至3.0),梯度洗脱程序见表1;流速:1.0 ml/min;进样量:20µl;检测波长:240 nm;柱温:25Ⅱ。

表1 梯度洗脱程序Tab 1 Gradient elution program
2.3 溶液的制备
2.3.1 对照品溶液 精密称取干燥至恒质量的Nap对照品10.0 mg,置于10 ml量瓶中,用甲醇溶解并稀释至刻度,摇匀,制得质量浓度为1 000µg/ml的Nap对照品溶液。
2.3.2 供试品溶液 精密称取干燥至恒质量的Nap-SLN 10.0 mg(以Nap计),置于10 ml量瓶中,用甲醇溶解并稀释至刻度,超声(功率:250 W,频率:40 kHz)处理30 min,滤过,准确量取续滤液1.2 ml,置于10 ml量瓶中,用甲醇稀释至刻度,摇匀,制得质量浓度为120µg/ml的Nap供试品溶液。
2.3.3 阴性对照溶液 取处方量的硬脂酸、大豆磷脂、牛黄胆酸钠,按“2.3.2”项下方法操作制成阴性对照溶液。
2.4 阴性干扰试验
分别量取“2.3”项下的对照品溶液、供试品溶液和阴性对照溶液各适量,按“2.2”项下色谱条件进样测定,记录色谱,详见图1。由图1可知,对照品和供试品均在10.210 min处出现吸收峰,而阴性对照在此处无吸收峰出现,表明阴性对照对主成分Nap的含量测定无干扰。
2.5 线性关系考察

图1 高效液相色谱图A.对照品溶液;B.供试品溶液;C.阴性对照溶液;1.NapFig 1 HPLC chromatogramsA.reference substance solution;B.test sample solution;C.negative con trol solution;1.naproxen
将“2.3.1”项下的对照品溶液分别用甲醇稀释制成质量浓度为0.2、1.0、5.0、20.0、40.0、80.0、120.0、160.0和180.0µg/ml的系列对照品溶液,分别按“2.2”项下色谱条件注入HPLC仪,进样测定,记录峰面积。以Nap的峰面积(y)为纵坐标、质量浓度(x,µg/ml)为横坐标进行线性回归,得回归方程y=39 213x+ 42 880(r=0.999 7)。结果表明,Nap检测质量浓度线性范围为0.2~180.0µg/ml。
2.6 精密度试验
精密吸取“2.5”项下质量浓度为160µg/ml的对照品溶液20µl,按“2.2”项下色谱条件连续进样6次,记录峰面积。结果,Nap峰面积的RSD=0.65%(n=6),说明仪器精密度良好。
2.7 稳定性试验
取按“2.3.2”项下方法制备的供试品溶液(批号:20150801)适量,分别于室温下放置0、2、4、8、12、24 h时取20 µl,按“2.2”项下色谱条件进样测定,记录峰面积。结果,Nap峰面积的RSD=0.28%(n=6),说明供试品溶液在室温放置24 h内稳定性良好。
2.8 重复性试验
取同一批(批号:20150801)样品适量,共6份,分别按“2.3.2”项下方法制备供试品溶液,再按“2.2”项下色谱条件进样测定,记录峰面积。结果,Nap峰面积的RSD=0.84%(n=6),说明本方法重复性良好。
2.9 加样回收率试验
精密称取已知含量的样品(批号:20150801)7.5 mg,共9份,分别置于量瓶中,对照品按80%、100%、120%的量各加3份,按“2.3.2”项下方法制备供试品溶液,再按“2.2”项下色谱条件进样测定,记录峰面积并计算加样回收率,结果见表2。
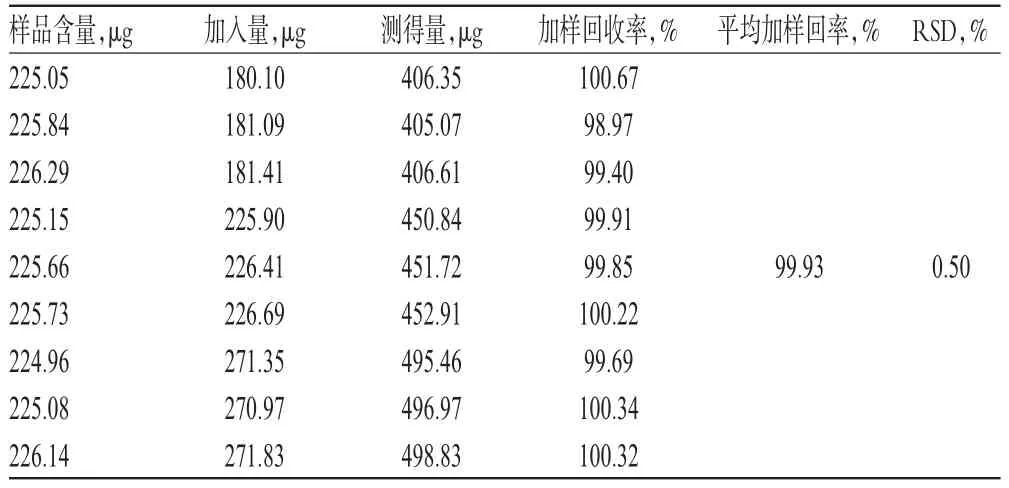
表2 加样回收率试验结果(n=9)Tab 2 Results of recovery tests(n=9)
2.10 样品含量测定
取3批样品各适量,分别按“2.3.2”项下方法制备供试品溶液,再按“2.2”项下色谱条件进样测定,记录峰面积并按外标法计算主成分的含量,结果见表3。

表3 样品含量测定结果(n=3,%)Tab 3 Results of contents determination of samples(n=3,%)
3 讨论
3.1 测定波长的选择
参考2015年版《中国药典》(二部)“萘普生”[10]项下的色谱条件,选择流动相A为甲醇、流动相B为0.01 mol/L磷酸二氢钾缓冲溶液(磷酸调节pH至3.0)(75∶25,V/V),分别采用药典中的Nap紫外吸收波长262、272、317和331 nm用于测定Nap-SLN时,可能由于辅料的干扰使Nap的峰形、保留时间均不理想。而Haque T等[11]曾报道了在240 nm波长下对Nap和盐酸雷尼替丁混合制剂进行分析,故本试验尝试采用该波长测定Nap-SLN中主成分Nap的含量。结果表明,在该波长下Nap-SLN中的辅料不干扰主成分的测定,峰形、保留时间均较理想。
3.2 流动相梯度洗脱程序的确定
有机相在整个流动相中所占比例对灵敏度、分离度以及分离时间均有明显的影响。为了使Nap-SLN中主成分Nap与辅料有效分离,本试验对梯度洗脱程序进行了考察。结果,确定了最佳梯度洗脱程序:甲醇初始体积为50%,恒定1 min,在1~2 min时由50%增加至65%,在2~5 min时由65%增加至70%,在5~7 min时由70%增加至90%,在7~10 min时由90%减少至50%,再恒定5 min。通过此梯度洗脱程序可使Nap保留时间适中,峰形较好,分离度符合要求。
3.3 样品含量测定结果分析
本试验建立了新的测定Nap-SLN中主成分含量的HPLC法,从含量测定结果可知,各批次的Nap-SLN中Nap含量无明显差异。试验结果表明,本方法简便、精确,可排除Nap-SLN中辅料的干扰,适用于其中主成分的含量测定。为该制剂包封率检测等理化性质评价及活性测定等相关后续研究奠定了方法学基础。
[1] 张伦.萘普生市场透析[J].中国药房,2003,14(6):326.
[2] 李鹏,田青平,李菁.萘普生微乳经皮给药系统的处方前研究[J].中国新药杂志,2008,17(22):1 948.
[3] Harde H,Das M,Jain S.Solid lipid nanoparticles:an oral bioavailability enhancer vehicle[J].Expert Opin Drug Deliv,2011,8(11):1 407.
[4] 夏爱晓,宋倩倩,孙渊.固体脂质纳米粒制备及应用研究进展[J].药学实践杂志,2012,30(5):331.
[5] Madan J,Pandey RS,Jain V,et al.Poly(ethylene)-glycol conjugated solid lipid nanoparticles of noscapine improve biological half-life,brain delivery and efficacy in glioblastoma cells[J].Nanomedicine,2013,9(4):492.
[6] 毛世瑞,王燕芝,纪宏宇,等.微乳化技术制备固体脂质纳米粒[J].药学学报,2003,38(8):624.
[7] 詹国平,潘道丽.紫外分光光度法测定萘普生-β-环糊精包合物中萘普生的含量[J].中国药房,2006,17(15):1 175.
[8] Zakeri-Milani P,Barzegar-Jalali M,Tajerzadeh H,et al. Simultaneous determination of naproxen,ketoprofen and phenol red in samples from rat intestinal permeability studies:HPLC method development and validation[J].J Pharm Biomed Anal,2005,39(3/4):624.
[9] Marengo E,Cavalli R,Caputo O,et al.Scale-up of the preparation process of solid lipid nanospheres.part I[J].Int J Pharm,2000,205(1/2):3.
[10] 国家药典委员会.中华人民共和国药典:二部[S].2015年版.北京:中国医药科技出版社,2015:1 208-1 209.
[11] Haque T,Talukder MM,Laila S,et al.Development and validation of a RP-HPLC method for simultaneous estimation of naproxen and ranitidinehydrochloride[J].Pak J Pharm Sci,2010,23(4):379.
Content Determination of Main Ingredient in Naproxen Solid Lipid Nanoparticles by HPLC
FU Jia,GAO Sainan,ZHOU Xuegang,XIN Ping,SUN Shiqin[College of Pharmacy,Harbin Medical University(Daqing Area),Heilongjiang Daqing 163319,China]
OBJECTIVE:To establish a method for the content determination of main ingredient in naproxen solid lipid nanoparticles.METHODS:Microemulsion method was used to prepare naproxen solid lipid nanoparticles,and HPLC was used for the content determination.The column was Elite C18with mobile phase A of methanol and B of 0.01mol/L KH2PO4buffer solution(adjust pH to 3.0 by phosphoric acid)(gradient elution)at a flow rate of 1.0 ml/min,detection wavelength was 240 nm,column temperature was 25Ⅱ,and injection volume was 20 μl.RESULTS:The linear range of naproxen was 0.2-180.0µg/ml(r=0.999 7);RSDs of precision,stability and reproducibility were lower than 1.00%;recovery was 99.73%-100.29%(RSD=0.20%,n=9). CONCLUSIONS:The method is simple,accurate,and can exclude the accessories’interference in naproxen solid lipid nanoparticles,which is suitable for the content determination of main ingredient.
Naproxen solid lipid nanoparticles;HPLC;Content determination
R927.2
A
1001-0408(2016)36-5127-03
2015-12-30
2016-11-09)
(编辑:周 箐)
黑龙江省自然科学基金资助项目(No.C2007-01);大庆市指导性科技计划项目(No.szd-2015-12)
*硕士。研究方向:类风湿性关节炎。E-mail:13159826215@ 163.com
#通信作者:教授,博士。研究方向:类风湿性关节炎。电话:0459-8153631。E-mail:ssq7610@126.com
DOI10.6039/j.issn.1001-0408.2016.36.27
