Identifcation of Risk Pathways and Functional Modules for Coronary Artery Disease Based on Genome-wide SNP Data
2016-03-09XiangZhaoYiZhaoLuanXiaoyuZuoYeDaChenJihengQinLvJinYiqingTanMeihuaLinNaizunZhangYanLiangShaoQiRao
Xiang ZhaoYi-Zhao LuanXiaoyu ZuoYe-Da Chen Jiheng QinLv JinYiqing TanMeihua LinNaizun Zhang Yan LiangShao-Qi Rao*k
1Institute for Medical Systems Biology and Department of Medical Statistics and Epidemiology,School of Public Health,Guangdong Medical College,Dongguan 523808,China
2School of Life Sciences,Sun Yat-sen University,Guangzhou 510080,China
3Department of Medical Statistics and Epidemiology,School of Public Health,Sun Yat-sen University,Guangzhou 510080,China
4Maoming People’s Hospital,Maoming 525000,China
Identifcation of Risk Pathways and Functional Modules for Coronary Artery Disease Based on Genome-wide SNP Data
Xiang Zhao1,#,a,Yi-Zhao Luan2,#,b,Xiaoyu Zuo3,c,Ye-Da Chen1,d, Jiheng Qin1,e,Lv Jin1,f,Yiqing Tan1,g,Meihua Lin1,h,Naizun Zhang3,i, Yan Liang4,j,Shao-Qi Rao1,3,*,k
1Institute for Medical Systems Biology and Department of Medical Statistics and Epidemiology,School of Public Health,Guangdong Medical College,Dongguan 523808,China
2School of Life Sciences,Sun Yat-sen University,Guangzhou 510080,China
3Department of Medical Statistics and Epidemiology,School of Public Health,Sun Yat-sen University,Guangzhou 510080,China
4Maoming People’s Hospital,Maoming 525000,China
Coronary artery disease;
Genome-wide SNP profling;
Risk pathway;
Functional module;
Genetic network
Coronary artery disease(CAD)is a complex human disease,involving multiple genes and their nonlinear interactions,which often act in a modular fashion.Genome-wide single nucleotide polymorphism(SNP)profling provides an effective technique to unravel these underlying genetic interplays or their functional involvements for CAD.This study aimed to identify the susceptible pathways and modules for CAD based on SNP omics.First,the Wellcome Trust Case Control Consortium(WTCCC)SNP datasets of CAD and control samples were used to assess the jointeffect of multiple genetic variants at the pathway level,using logistic kernel machine regression model.Then,an expanded genetic network was constructed by integrating statistical gene-gene interactions involved in these susceptible pathways with their protein-protein interaction(PPI) knowledge.Finally,risk functional modules were identifed by decomposition of the network.Of 276 KEGG pathways analyzed,6 pathways were found to have a signifcant effect on CAD.Other than glycerolipid metabolism,glycosaminoglycan biosynthesis,and cardiac muscle contraction pathways,three pathways related to other diseases were also revealed,including Alzheimer’s disease,non-alcoholic fatty liver disease,and Huntington’s disease.A genetic epistatic network of 95 genes was further constructed using the abovementioned integrative approach.Of 10 functional modules derived from the network,6 have been annotated to phospholipase C activity and cell adhesion molecule binding,which also have known functional involvement in Alzheimer’s disease. These fndings indicate an overlap of the underlying molecular mechanisms between CAD and Alzheimer’s disease,thus providing new insights into the molecular basis for CAD and its molecular relationships with other diseases.
Introduction
Coronary artery disease(CAD)is the leading cause of morbidity and mortality worldwide and has a strong genetic basis[1]. Advances in genome-wide association studies(GWAS)have provided insights into lots of different genetic factors that contribute to the disease.Since 2007,the Wellcome Trust Case Control Consortium(WTCCC)and Framingham Heart Study have achieved duplicated validation of 9p21,and identifcation of 13 novel loci associated with CAD[2,3].More similar studies have also been performed in different populations,leading to the excavation of more CAD-related single nucleotide polymorphisms(SNPs)[4,5].As a result,population-based studies with thousands of patients and healthy controls included have identifed more than 50 CAD-associated genetic loci in total [6-9].
However,all these studies did not take into account the underlying genetic interplays or functional modules involved in CAD.Therefore,the genetic basis of CAD has remained largely unknown due to the limited understanding of just a small proportion of individual genetic variations.There is a developing consensus that genetic variations of CAD often function by sophisticated interactions through a modular fashion rather than individually[10].With the improvement of computational methods,gene set-based association analysis(GSA) aimed at evaluating the joint effects of a defned gene set by quantifying the susceptibility or statistical signifcance of individual functional units,e.g.,pathways or biological processes, associated with clinical phenotypes[11].
In general,the gene sets in GSA can be generated according to the manually-curated pathways(e.g.,KEGG pathways),or gene lists related to specifc functions.Based on the differences in the theoretical models used,the pathway-based methods can be classifed into three categories,i.e.,functional enrichment analysis[12,13],topology-based analysis[14],and multivariate statistical analysis.Hereinto,functional enrichment analysis is the most widely used method for the identifcation of enriched pathways related to the phenotype of interest due to its straightforward statistical model.For instance,Ghosh et al. [15]applied Reactome gene sets-based gene set enrichment analysis(GSEA)to reveal novel associations between key biological processes and CAD.By contrast,topology-based methods were proposed based on the fact that biological pathways are not simple lists of genes,but rather complex interactions between genes with specifc topology.A list of topologybased methods and their applications have been reviewed previously[16].However,so far,such methods are largely under development and their applications on CAD have rarely been reported.Instead,logistic kernel machine regression[17,18],a semi-parametric regression model,is often used as the theoretical framework for multivariate analysis of multiple genetic variants in a gene set.It can be used to examine the signifcance of a gene set that corresponds to specifc functional units such as pathways.By mapping the feature vector of genes or SNPs to a phenotype similarity space that indicates the similarity between people carrying these genes or SNPs,the logistic kernel machine regression analysis considers the linear interactions among the genes and thus shows advantage over the classical GSEA,
In this article,we report the fndings from genome-wide and pathway-based analysis of a publicly-available GWAS dataset provided by WTCCC.We frst systematically assessed the association of each KEGG pathway with CAD using the logistic kernel regression model.Then,we constructed an expanded genetic network by integrating gene-gene interactions involved in these susceptible pathways with their protein-protein interactions(PPIs).Finally,we identifed the risk functional modules(subnetworks)for CAD by decomposing the built genetic network.
Results
SNP set test identifed CAD susceptible KEGG pathways
The raw WTCCC genotyping data for CAD contained 482,247 SNPs from 5000 individuals(3000 controls and 2000 CAD patients).After data processing,101,822 SNPs from 4864 individuals(2938 controls and 1926 CAD patients)were retained for further analysis.We defned the SNPs annotated to the genes of the same KEGG pathway as a SNP set.Hence,276 pathway-based SNP sets were generated in total.These SNP sets were tested by logistic kernel machine regression model to evaluate whether they jointly(i.e.,pathway-based)contribute signifcantly to CAD risk.As a result,totally 6 pathways were identifed to be signifcantly associated with CAD(Bonferroni-adjusted P<0.05;Table 1).These includeglycerolipid metabolism,glycosaminoglycan biosynthesis,and cardiac muscle contraction,as well as three disease-related pathways,i.e.,Alzheimer’s disease(AD),non-alcoholic fatty liver disease(NAFLD),and Huntington’s disease.

Table 1 Signifcant susceptible pathways for CAD
Interestingly,potential correlation between some of these pathways and CAD has been reported in previous studies. For example,de las Fuentes et al.[19]performed a pathwaybased analysis of another independent GWAS dataset for the Framingham Heart Study and identifed glycerolipid metabolism pathway to be signifcantly associated with CAD.In agreement with this study,a previous report showed that the level of serum triglyceride,a key element in glycerolipid metabolism,could be used as an effective predictor for CAD risk [20].There is also evidence showing relevance of glycosaminoglycan biosynthesis with CAD.Glycosaminoglycan is present extensively in various cell types,to maintain the resilience and permeability of vascular wall or play a key role in anticoagulation and antihyperlipidemia[21].Therefore,genetic variation in this pathway may cause dysfunction of blood vessels. It is not surprising that cardiac muscle contraction(hsa04260) was identifed to be the risk pathway for CAD.Myocardial contraction is an initial process of potential shift of cardiac muscle cells,to produce longitudinal,radical,and rotational motion.Li et al.[22]applied an ultrasonic imaging technique and showed that cardiac muscle motion of CAD patients is signifcantly different from that of health subjects at various directions,implying the correlation between myocardial dysfunction and myocardial contraction in CAD patients.
Notably,our study revealed that three pathways related to other diseases were linked with CAD.It has been reported that among all kinds of fatty liver diseases examined,NAFLD shows the strongest correlation with CAD[23],and NAFLD patients have a higher risk for cardiovascular disease[24]. The correlation between AD,a chronic neurodegenerative disease,and CAD was also noticed because of occurrence of cognitive impairment in CAD patients[25].Nevertheless,there is dearth of evidence supporting the relevance of CAD with Huntington’s disease,a Mendelian neurodegenerative disorder with autonomic dominant inheritance.
Taken together,most of the pathways identifed in this study have clear evidence supporting their involvement in the underlying pathogenesis for CAD.
CAD-related genetic network analysis identifed PIK3R1 and APP as hub genes
Epistasis analysis of all SNP-SNP pairs within or across the identifed pathways was performed.Totally 186,640 SNP-SNP signifcant interactions(P<0.05)were identifed. We then mapped the involved SNPs onto genes.By integrating prior PPI knowledge,we constructed genetic networks using 121 unique genes and 149 gene-gene pairs.As shown in Figure 1,most of these genes were connected to each other, producing the largest sub-network with 95 unique genes and 135 edges.There were also 9 small sub-networks including seven sub-networks with only one edge,one sub-network with two edges,and one sub-network with three edges.Furthermore,there are three genes that were not linked to any other genes at all due to the lack of PPI evidence.
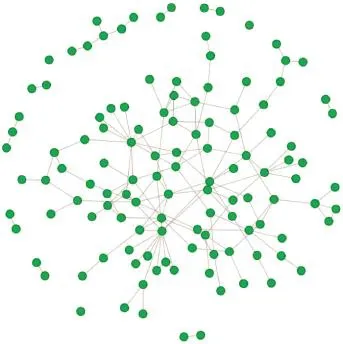
Figure 1 Epistatic network for CAD
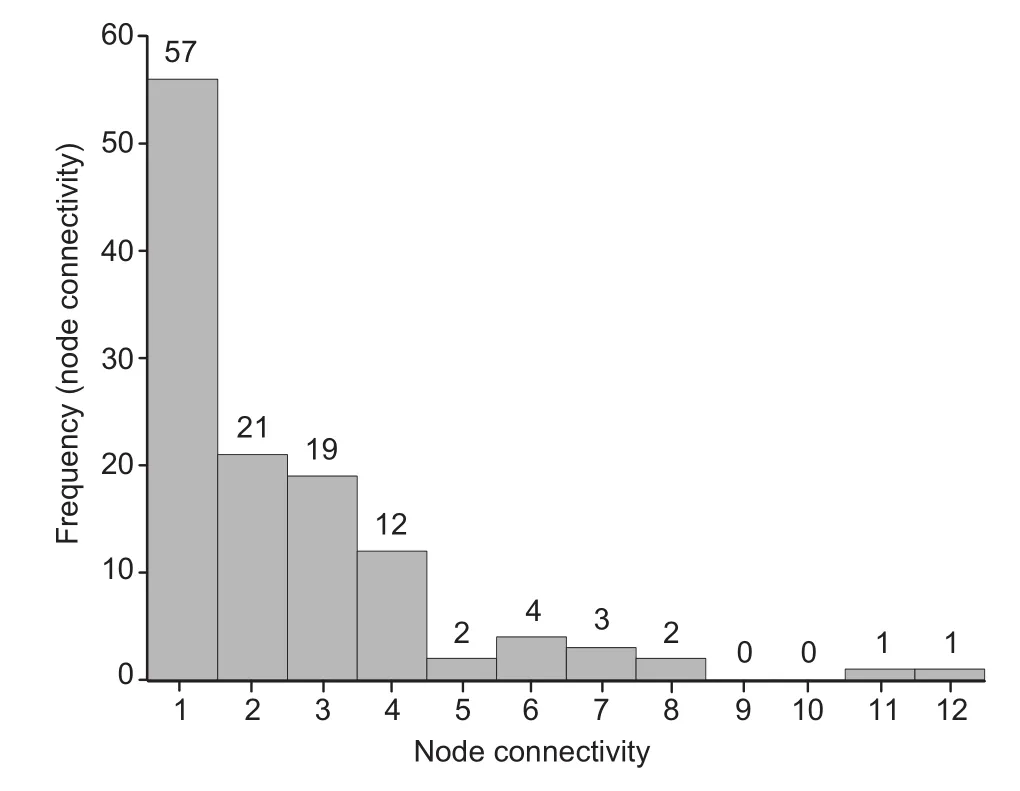
Figure 2 Frequency distribution of node connectivity for the largest genetic network of CAD
We focused on the largest sub-network for the following topological analysis.The connection degree distribution of the largest sub-network(Figure 2)indicated that this network is not a random network.Furthermore,Kolmogorov-Smirnov (KS)test[26]showed that this network was a scale-free network with an exponential parameter α=3.023 (P=0.884).Among all the genes,two genes,PIK3R1 and APP,showed the highest connectivity,which are connected with 11(Bonferroni-adjusted P=0.0041)and 12 other genes (Bonferroni-adjusted P=0.00088),respectively.These two genes were thus defned as the hub genes.
PIK3R1 encodes the regulatory subunit α of phosphatidylinositol 3-kinase(PIK3),which orchestrates a series of cell function regulation,such as cell proliferation,cell differentiation,apoptosis,and glucose transport[27].PIK3 can be activated by angiotensin II,and the activated PIK3 plays a vital role in vascular smooth muscle cells through angiotensin II stimulated Ca2+entry[28].APP,which encodes β-amyloid precursor protein,is generally recognized to be closely related to AD[29].Abnormal expression of APP can lead to dysfunction of endothelial cells due to cytotoxicity and damage induced by long-term exposure to Aβ peptide[30,31].These studies provide supporting evidence of PIK3R1 and APP on the development of CAD.
CAD-related genetic network module analysis reveals the involvement of various molecular functions
To identify the most compact functional subnetworks,we further decomposed the largest network into smaller modular units.We obtained totally 10 modules,which consist of 4-14 genes(Figure 3).The corresponding gene lists can be found in Table S1.Interestingly,KS test revealed that all modules were scale-free with P values ranging 0.8-1.0,except the smallest one which only contained 4 genes(Table 2).The estimates of the scaling exponent(α)of power law distribution,KS testing statistics,and some other topological properties for each module are also shown in Table 2.
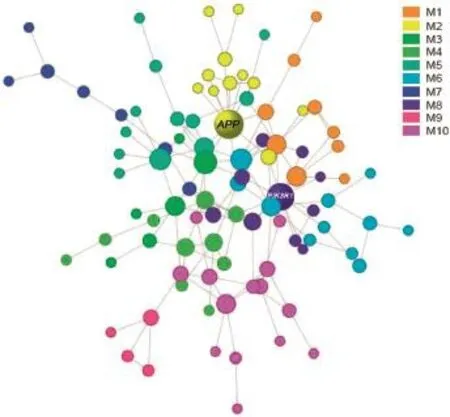
Figure 3 Modular partitions of CAD risk genes
To understand the function involvements of each module, we performed a gene ontology(GO)-based enrichment analysis using the database for annotation,visualization and integrated discovery(DAVID).Only modules containing more than 10 genes were analyzed and the signifcantly-enriched GO terms for each module are listed in Table 3.We found that different modules had some characteristic functional involvements.For examples,M3 and M5 were signifcantly enriched with the cellular component of cytosol,M9 with cell nucleus,and M6 with neuron related structures,indicating that these modules had very different‘working places’.In addition,these modules also exhibited varied molecular functions.For instances,M3 was linked to the lipid-related functions,M2 to peptidase activity, and M6 to cell adhesion molecule binding activity.Notably, M6 was also enriched with biological process neuronal activity,which could be the molecular bridge between AD and CAD.
Among all these enriched GO terms,phospholipase C activity(GO:0004629)and cell adhesion molecule binding(GO: 0050839)took special attention.Phospholipase C(PLC), which is distributed widely in various tissues,is a key enzyme in phosphatidylinositol signaling pathway[31].There exist different isozyme types of PLC,including PLC-β,PLC-γ,and PLC-δ.Theseisozymespossessconserved and specifc domains,and therefore PLC activation may be induced in various ways[32].PLC-γ is mostly activated through protein tyrosine kinases(PTKs).As a result,activated PLC-γ would induce a series of signal transduction,which may lead to trans-activation of epidermal growth factor(EGF),a key element in inhibition of vascular wall deposition and thrombogenesis [33,34]. Additionally, by mediating inter-cellular interactions,cell adhesion molecules(CAMs)can regulate multiple biological processes,such as signal transduction, infammation and immune responses,coagulation and tissue repairing[35].The changes in adhesion of vascular endothelial cellsurface,e.g.,caused by injury,promotemonocyteadhesion.During the migration and transformation to macrophages,monocyte may stimulate lymphocytes to produce multiple cytokines,thus promoting proliferation of smooth muscle and formation of fbrous plaque eventually[36].
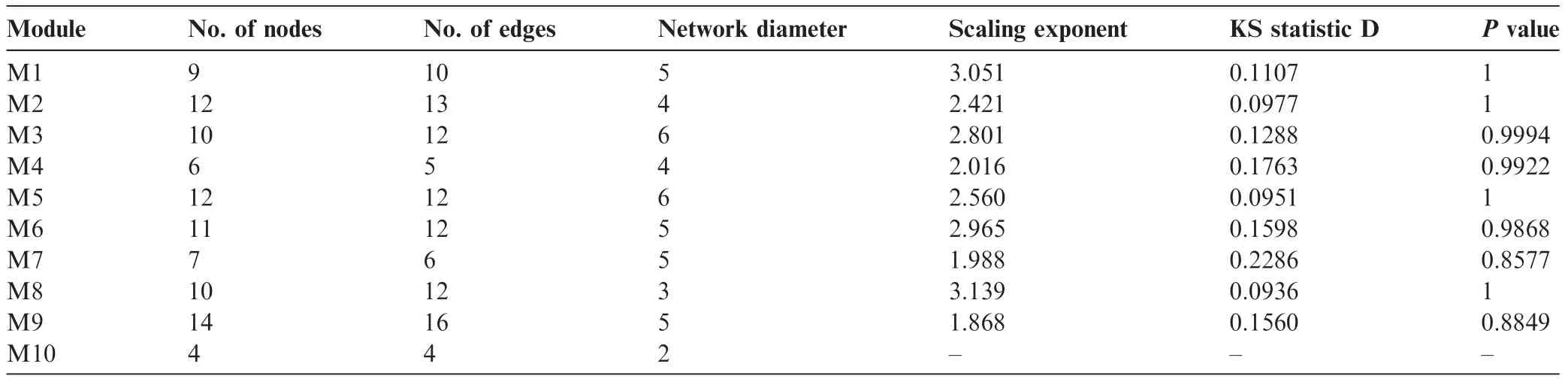
Table 2 The topological features of the risk modules for CAD

Table 3 The GO terms enriched for each risk module for CAD
Discussion
Studies of genetic variants in various molecular biological processes will greatly promote our understanding of CAD and its pathogenesis.In the present study,a pathway-based approach is applied to identify the risk pathways and functional modules for CAD.This study demonstrated that multiple pathways may be involved in the underlying molecular processes for CAD,such as cardiac muscle contraction,glycerolipid metabolism,and glycosaminoglycan biosynthesis. Meanwhile,this study reveals that some other diseases,e.g., non-alcoholic fatty liver disease,Huntington’s disease,and AD,may share molecular mechanisms with CAD.Furthermore,2 hub genes,PIK3R1 and APP,and 6 risk functional modules for CAD have been identifed.Our fndings are different from Liu’s study[37].Using gene co-expression network analysis,Liu et al.identifed specifc modules and hub genes that are mainly related to membrane-associated processes and hypertrophic cardiomyopathy pathway.The possible reasons for the difference may be manifold,one of which is the difference in the choice of omics data type.They evaluated gene expression changes at mRNA level,whereas we analyzed the genomic mutations within DNA sequences. Overall,our study provides new insights into the molecular basis for CAD and its molecular relationships with several related diseases.
Genetic studies are traditionally based on single gene analysis,which poses tremendous challenges for elucidating complicated genetic interplaysinvolved in complex human diseases.Modern pathway-based analysis allows a comprehensive understanding of the molecular mechanisms underlying complex diseases by considering the joint effect and integrality as function unit of multiple genes.Extensive studies utilizing pathway-based analysis have signifcantly advanced our capacity to explore large-scale omics data that have been rapidly accumulating in biomedical felds[38].
Pathway-based approach has some advantages over the conventionalGWASapproach.Thesinge-locusanalysis widely used in GWAS is only capable of capturing a small portion of susceptible SNPs with prominent marginal effects,leaving the important genetic component,such as epistasis or joint effects between multiple genes,undetected.Identifying the complex interplays among multiple genes in the genome-wide context is an essential task to systematically unravel the molecular mechanisms underlying complex diseases.In this study, we employed the newly-developed logistic kernel machine regression model in the pathway-level analysis to capture the joint effects of multiple genes involved in the pathways.In this way,we are not only able to avoid the curse of‘high dimensionality and small sample size’associated with analysis of GWAS data,but also able to estimate the missing genetic components and epistasis,thus helping elucidate the sophisticated molecular interplays between or across the risk pathways for CAD.
There are also limitations in this study.First,the whole pathogenic process of CAD may involve a long cascade of multiple biological pathways.Ideally,the accumulative effects of these risk pathways on CAD should be examined,which is practically diffcult to achieve because of the unpredictability of joint action among different pathways and the possibility of over-ftting.Second,we replaced missing genotypes with the most frequent allele.Such simplifcation for data imputation could lead to expansion of major alleles,resulting in a decreased minor allele frequency(MAF).On the other hand, genotype imputation is a complex process in GWAS research, which can address the failures occurring during genotyping assay to some extent[39,40].However,in practice,most imputation methods require external reference panel of SNPs that may introduce noise of genetic background,and the success of imputation is largely determined by the patterns of linkage disequilibrium(LD)[41].In our analysis,the missing genotypes were imputed after fltering SNPs and subjects with missing rate≥5%,which help restrain the expansion of major alleles. Moreover,a gene-set-based method rather than a singlelocus-based method was applied in our further analysis,reducing the bias of single SNPs resulting from the imputation. Third,in the epistatic analysis,we used the nominal P value of 0.05 to identify the putative epistatic gene pairs,which could lead to an infated type I error.Correction for multiple tests is a very complicated and challenging issue for the analysis of largescale GWAS data,especially for epistatic analysis because of correlation of gene pairs or correlations stemming from LD. To address this concern,we applied an additional criterion of experimentally-confrmed PPI support for gene pairs,which might alleviate the issue of the infated type I errors to some extent.Finally,this study only integrated GWAS data with PPI data for genetic network analysis.Integrating more omics data such as epigenetic or epidemiological data would help illustrate the genetic,epigenetic,and environmental factors for CAD,which is the focus in our future studies.
Materials and methods
Data sources
WTCCC genotyping data of 482,247 SNPs from 2000 CAD subjects and 3000 health control subjects[42]were analyzed in this study.For identifcation of susceptible pathways,283 human pathways[43,44]were extracted and downloaded from KEGG database.We removed the more general pathways that contain several specifc pathways,and ultimately included 276 KEGG pathways.Genetic information for mapping SNPs to genes was extracted from Ensembl/GRCh37[16].Information on PPIs was retrieved from the Human Protein Reference Database(HPRD)[45]for genetic networking(epistatic interactions).To enhance the reliability of the depicted genetic relationships,only experimentally-confrmed PPIs were taken as the prior knowledge.
Data preprocessing
To improve the data quality,several data preprocessing procedures were performed.First,SNPs were excluded if they did not meet all the following criteria:(1)genotype missing rate<5%,(2)individualmissing rate<5%,and (3) MAF>0.01.Furthermore,all the included loci must meet Hardy-Weinberg equilibrium (HWE) proportions (P>1×10-4)in the control group.Second,for loci with missing values after the fltering above,missing genotypes were replaced with the most frequent one.Third,to remove data redundancy due to LD,only tag SNPs that were representative in the corresponding genomic regions were utilized for the current analysis.For identifcation of these tag SNPs,each individual chromosome was scanned using the‘moving window’method in which the window size was set to 50 SNPs with step length of 5 SNPs.The cutoff of LD r2was set as 0.8.Finally, SNPs were considered as mapped onto genes if these SNPs are situated in the fanking regions spanning from 5 kb upstream to 5 kb downstream of the genes,as described previously [46].All the data processing procedures were performed using PLINK program[47]and R platform(http://www.r-project. org/).
Identifcation of CAD susceptible pathways
Logistic kernel machine regression model was applied to identify the susceptible pathways related to CAD.Suppose that a pathway contains p SNPs(their genotypes are denoted as zi*) and P(yi=1)be the probability of the ith subject being affected(i.e.,who has the disorder).This model can be described as follows:
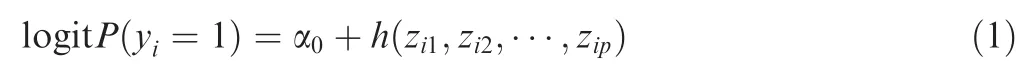
where α0is the intercept,and h(·)is a general function of p SNPs contained in the pathway,which is often defned as a positive,semi-defnite kernel function K(·,·).In this study,this kernel function is defned as

where the weight wiis calculated as described previously[48]. More intuitively,K(.,.)can be viewed as a function that measures the similarity between two individuals based on the genotypes of the SNPs in the SNP set.There are three options for K (.,.):the linear,Gaussian,and identical-by-state kernels.The null hypothesis for testing a pathway is that its overall effect is zero,i.e.,H0:h(·)=0.The signifcance of the pathwaybased SNP set was tested by Q statistics that follows a χ2mixeddistribution.To adjust for multiple pathways to be evaluated, Bonferronicorrection was applied and signifcancewas claimed if P×N<0.05(N is the number of pathways evaluated).More details about the methods used are described previously[48,49].
Genetic networking
Function enrichment analysis
To characterize the functional involvement of the putative modules for CAD,GO analysis was performed for each module using DAVID[51,52]with the whole human genome genes as background,and the gene list within each module as foreground.To control false positive rate of signifcance of GO terms,Bonferroni correction was used.In order to better characterize the putative modules,we reported the GO terms with node depth≥4.The information for GO hierarchy was retrieved from Bioconductor GO.db and the node depth for each GO term was defned by the minimum distance between target GO term and GO root term in the tree structure.
Authors’contributions
SQR conceived the project and wrote the manuscript.XZ and YZL performed the analysis and wrote the manuscript.XZ, YDC,JQ,LJ,YT,ML,NZ,YL participated in writing the computing codes and analyzing the public datasets.All authors read and approved the fnal manuscript.
Competing interests
The authors declare no competing interests.
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China(Grant Nos.31071166 and 81373085),Natural Science Foundation of Guangdong Province,China(Grant No.8251008901000007),Science and Technology Planning Project of Guangdong Province(Grant No.2009A030301004),Dongguan Science and Technology Project,Guangdong,China(Grant No.2011108101015),and the funds from Guangdong Medical College,China(Grant Nos.XG1001,JB1214,XZ1105,STIF201122,M2011024, and M2011010).
Supplementary material
Supplementary material associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j. gpb.2016.04.008.
[1]Albert R,Baraba´si AL.Statistical mechanics of complex networks.Rev Mod Phys 2002;74:47.
[2]Samani NJ,Erdmann J,Hall AS,Hengstenberg C,Mangino M, Mayer B,et al.Genomewide association analysis of coronary artery disease.N Engl J Med 2007;357:443-53.
教材是语言教学的载体,教师应该深挖教材内容且运用得当,以点带面,对比中西方文化差异,去粗取精,借鉴西方优秀文化,弘扬中国传统文化,营造浓厚的育人氛围。例如,我们在基础英语部分的教学重视对学生的人文素质教育,帮助学生初步了解中西文化的相异之处,并进行品德、心理、审美等教育,提高学生文化涵养;行业英语部分突出职业人文素质与专业结合的特色,培养学生高尚的职业道德和职业观念。
[3]Larson M,Atwood L,Benjamin E,Cupples LA,D’Agostino R, Fox C,et al.Framingham Heart Study 100K project:genomewide associations for cardiovascular disease outcomes.BMC Med Genet 2007;8:S5.
[4]Barbalic M,Reiner AP,Wu C,Hixson JE,Franceschini N,Eaton CB,et al.Genome-wide association analysis of incident coronary heart disease(CHD)in African Americans:a short report.PLoS Genet 2011;7:e1002199.
[5]Polfus LM,Smith JA,Shimmin LC,Bielak LF,Morrison AC, Kardia SL,et al.Genome-wide association study of gene by smoking interactions in coronary artery calcifcation.PLoS One 2013;8:e74642.
[6]Erbilgin A,Civelek M,Romanoski CE,Pan C,Hagopian R, Berliner JA,et al.Identifcation of CAD candidate genes in GWAS loci and their expression in vascular cells.J Lipid Res 2013;54:1894-905.
[7]Peden JF,Farrall M.Thirty-fve common variants for coronary artery disease:the fruits of much collaborative labour.Hum Mol Genet 2011;20:R198-205.
[8]Roberts R,Stewart AF.Genes and coronary artery disease:where are we?J Am Coll Cardiol 2012;60:1715-21.
[9]Gao H,Li L,Rao S,Shen G,Xi Q,Chen S,et al.Genome-wide linkage scan identifes two novel genetic loci for coronary artery disease:in genequest families.PLoS One 2014;9:e113935.
[10]Li H,Zuo X,Ouyang P,Lin M,Zhao Z,Liang Y,et al. Identifying functional modules for coronary artery disease by a prior knowledge-based approach.Gene 2014;537:260-8.
[11]Fridley BL,Biernacka JM.Gene set analysis of SNP data: benefts,challenges,and future directions.Eur J Hum Genet 2011;19:837-43.
[12]Wang K,Li M,Bucan M.Pathway-based approaches for analysis ofgenomewide association studies.Am J Hum Genet 2007;81:1278-83.
[13]Barry WT,Nobel AB,Wright FA.Signifcance analysis of functional categories in gene expression studies:a structured permutation approach.Bioinformatics 2005;21:1943-9.
[14]Draghici S,Khatri P,Tarca AL,Amin K,Done A,Voichita C, et al.A systems biology approach for pathway level analysis. Genome Res 2007;17:1537-45.
[15]Ghosh S,Vivar J,Nelson CP,Willenborg C,Segre`AV,Ma¨kinen VP,et al.Systems genetics analysis of genome-wide association study reveals novel associations between key biological processes and coronary artery disease.Arterioscler Thromb Vasc Biol 2015;20:2081-6.
[16]Mitrea C,Taghavi Z,Bokanizad B,Hanoudi S,Tagett R,Donato M,et al.Methods and approaches in the topology-based analysis of biological pathways.Front Physiol 2013;4:278.
[17]Liu D,Ghosh D,Lin X.Estimation and testing for the effect of a genetic pathway on a disease outcome using logistic kernel machine regression via logistic mixed models.BMC Bioinformatics 2008;9:292.
[18]Wu MC,Kraft P,Epstein MP,Taylor DM,Chanock SJ,Hunter DJ,et al.Powerful SNP-set analysis for case-control genome-wide association studies.Am J Hum Genet 2010;86:929-42.
[19]de las Fuentes L,Yang W,Da´vila-Roma´n VG,Gu CC.Pathwaybased genome-wide association analysis of coronary heart disease identifes biologically important gene sets.Eur J Hum Genet 2012;20:1168-73.
[20]Manninen V,Tenkanen L,Koskinen P,Huttunen J,Ma¨ntta¨ri M, Heinonen O,et al.Joint effects of serum triglyceride and LDL cholesterol and HDL cholesterol concentrations on coronary heart disease risk in the Helsinki Heart Study:implications for treatment.Circulation 1992;85:37-45.
[21]Zhang YM,Li BC,Zhu LP,Dai WF,Fan JH.Research advancement on chemistry and bioactivities of natural polysaccharide. J Kunming Univ Sci Technol (Sci Technol) 2003;28:140-9.
[22]Yang L,Wu LM,Qiu Q.Investigation of ischemic myocardial systolic dysfunction in coronary artery disease patients with twodimensional strain imaging.Chin J Med Imaging Technol 2011;27:1392-5.
[23]Zhou Q,Gao FJ.Advances in research on relationship between non-alcoholic fatty liver and coronary heart disease.Chin J Arterioscler 2008;16:669-72.
[24]Gaggini M,Morelli M,Buzzigoli E,DeFronzo RA,Bugianesi E, Gastaldelli A.Non-alcoholic fatty liver disease(NAFLD)and its connection with insulin resistance,dyslipidemia,atherosclerosis and coronary heart disease.Nutrients 2013;5:1544-60.
[25]Stampfer M.Cardiovascular disease and Alzheimer’s disease: common links.J Intern Med 2006;260:211-23.
[26]Barabasi AL,Oltvai ZN.Network biology:understanding the cell’s functional organization.Nat Rev Genet 2004;5:101-13.
[27]Li X.Advance in research on phosphoinositide 3-kinase structure and function.J Chengdu Univ(Nat Sci Edn)2013;32:219-23.
[28]Macrez N,Mironneau C,Carricaburu V,Quignard JF,Babich A, Czupalla C,et al.Phosphoinositide 3-kinase isoforms selectively couple receptors to vascular L-type Ca2+channels.Circ Res 2001;89:692-9.
[29]Zhang YW,Thompson R,Zhang H,Xu H.APP processing in Alzheimer’s disease.Mol Brain 2011;4:3.
[30]Thomas T,Thomas G,McLendon C,Sutton T,Mullan M.β-Amyloid-mediated vasoactivity and vascular endothelial damage. Nature 1996;380:168-71.
[31]Suo Z,Fang C,Crawford F,Mullan M.Superoxide free radical and intracellular calcium mediate A beta(1-42)induced endothelial toxicity.Brain Res 1997;762:144-52.
[32]Rhee SG.Regulation of phosphoinositide-specifc Phospholipase C.Annu Rev Biochem 2001;70:281-312.
[33]Rong LL,Hai XS.Progress in the structure of phospholipase C and mechanisms of its activation.Acta Acad Med CPAPF 2006;15:175-8.
[34]Wang ZD,Ping ZL,Yuan ZZ.Expression and its signifcance of phospholipase C-γ1 in tumors.Int J Stomatol Occlusion Med 2012;39:775-7.
[35]Zhou T,Wang F,Sun GZ,Zhang YM.Some biology progress of the adhesion molecules and sugar adhere to their cell.J Chin Microcirc 2003;7:187-90.
[36]Liang P,Sun L,Tang JW,Wang C.Expression of intercellular adhesion molecule-1,vascular cell adhesion molecule-1,tumor necrosis factor-α in atherosclerosis immunohistochemistry examination.Chin J Arterioscler 2004;12:427-9.
[37]Liu J,Jing L,Tu X.Weighted gene co-expression network analysis identifes specifc modules and hub genes related to coronary artery disease.BMC Cardiovasc Disord 2016;16:54.
[38]Jin L,Zuo XY,Su WY,Zhao XL,Yuan MQ,Han LZ,et al. Pathway-based analysis tools for complex diseases:a review. Genomics Proteomics Bioinformatics 2014:210-20.
[39]Jiao S,Hsu L,Hutter CM,Peters U.The use of imputed values in the meta-analysis of genome-wide association studies.Genet Epidemiol 2011;35:597-605.
[40]Korte A,Farlow A.The advantages and limitations of trait analysis with GWAS:a review.Plant Methods 2013;9:227-35.
[41]Pei YF,Li J,Zhang L,Papasian CJ,Deng HW.Analyses and comparison of accuracy of different genotype imputation methods.PLoS One 2007;3:e3551.
[42]Burton PR,Clayton DG,Cardon LR,Craddock N,Deloukas P, Duncanson A,et al.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls.Nature 2007;447:661-78.
[43]Kanehisa M,Goto S.KEGG:Kyoto encyclopedia of genes and genomes.Nucleic Acids Res 2000;28:27-30.
[44]Kanehisa M,Goto S,Sato Y,Kawashima M,Furumichi M, Tanabe M.Data,information,knowledge and principle:back to metabolism in KEGG.Nucleic Acids Res 2014;42:D199-205.
[45]Prasad TK,Goel R,Kandasamy K,Keerthikumar S,Kumar S, Mathivanan S,et al.Human protein reference database-2009 update.Nucleic Acids Res 2009;37:D767-72.
[46]Torkamani A,Topol EJ,Schork NJ.Pathway analysis of seven common diseases assessed by genome-wide association.Genomics 2008;92:265-72.
[47]Purcell S,Neale B,Todd-Brown K,Thomas L,Ferreira MA, Bender D,et al.PLINK:a tool set for whole-genome association and population-based linkage analyses.Am J Hum Genet 2007;81:559-75.
[48]Wu MC,Lee S,Cai T,Li Y,Boehnke M,Lin X.Rare-variant association testing for sequencing data with the sequence kernel association test.Am J Hum Genet 2011;89:82-93.
[49]Davies RB.Algorithm AS 155:The distribution of a linear combination of χ2random variables.J R Stat Soc Ser C Appl Stat 1980;29:323-33.
[50]Newman ME.Modularity and community structure in networks. Proc Natl Acad Sci U S A 2006;103:8577-82.
[51]HuangDW,ShermanBT,LempickiRA.Systematicand integrative analysis of large gene lists using DAVID bioinformatics resources.Nat Protoc 2008;4:44-57.
[52]Huang DW,Sherman BT,Lempicki RA.Bioinformatics enrichment tools:paths toward the comprehensive functional analysis of large gene lists.Nucleic Acids Res 2009;37:1-13.
Received 21 February 2016;revised 30 March 2016;accepted 10 April 2016 Available online 11 December 2016
Handled by Andreas Keller
*Corresponding author.
E-mail:raoshaoq@gdmc.edu.cn(Rao SQ).
#Equal contribution.
aORCID:0000-0002-5831-7188.
bORCID:0000-0003-4396-6078.
cORCID:0000-0002-1200-1022.
dORCID:0000-0002-7979-3482.
eORCID:0000-0003-0803-2843.
fORCID:0000-0001-9968-856X.
gORCID:0000-0001-6647-5090.
hORCID:0000-0002-2744-3704.
iORCID:0000-0003-4470-5582.
jORCID:0000-0001-6290-5540.
kORCID:0000-0002-7955-0480.
Peer review under responsibility of Beijing Institute of Genomics,Chinese Academy of Sciences and Genetics Society of China.
http://dx.doi.org/10.1016/j.gpb.2016.04.008 1672-0229©2016 The Authors.Production and hosting by Elsevier B.V.on behalf of Beijing Institute of Genomics,Chinese Academy of Sciences and Genetics Society of China.
This is an open access article under the CC BY license(http://creativecommons.org/licenses/by/4.0/).
