Six psychotropics for pre-symptomatic & early Alzheimer’s (MCI), Parkinson’s, and Huntington’s disease modification
2016-02-09EdwardLauterbachProfessorEmeritusofPsychiatryandNeurologyMercerUniversitySchoolofMedicineMaconGAUSA
Edward C. LauterbachProfessor Emeritus of Psychiatry and Neurology, Mercer University School of Medicine, Macon, GA, USA
Six psychotropics for pre-symptomatic & early Alzheimer’s (MCI), Parkinson’s, and Huntington’s disease modification
Edward C. Lauterbach*
Professor Emeritus of Psychiatry and Neurology, Mercer University School of Medicine, Macon, GA, USA
How to cite this article:Lauterbach EC (2016) Six psychotropics for pre-symptomatic & early Alzheimer’s (MCI), Parkinson’s, and Huntington’s disease modification. Neural Regen Res 11(11):1712-1726.
Open access statement:This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
The quest for neuroprotective drugs to slow the progression of neurodegenerative diseases (NDDs), including Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD), has been largely unrewarding. Preclinical evidence suggests that repurposing quetiapine, lithium, valproate, fluoxetine, donepezil, and memantine for early and pre-symptomatic disease-modification in NDDs may be promising and can spare regulatory barriers. The literature of these psychotropics in early stage and pre-symptomatic AD, PD, and HD is reviewed and propitious findings follow. Mild cognitive impairment (MCI) phase of AD: salutary human randomized controlled trial findings for low-dose lithium and, in selected patients, donepezil await replication. Pre-symptomatic AD: human epidemiological data indicate that lithium reduces AD risk. Animal model studies (AMS) reveal encouraging results for quetiapine, lithium, donepezil, and memantine. Early PD: valproate AMS findings show promise. Pre-symptomatic PD: lithium and valproate AMS findings are encouraging. Early HD: uncontrolled clinical data indicate non-progression with lithium, fluoxetine, donepezil, and memantine. Pre-symptomatic HD: lithium and valproate are auspicious in AMS. Many other promising findings awaiting replication (valproate in MCI; lithium, valproate, fluoxetine in pre-symptomatic AD; lithium in early PD; lithium, valproate, fluoxetine in pre-symptomatic PD; donepezil in early HD; lithium, fluoxetine, memantine in pre-symptomatic HD) are reviewed. Dose- and stage-dependent effects are considered. Suggestions for signal-enhancement in human trials are provided for each NDD stage.
drug repositioning; neuroprotective agents; psychotropic drugs; neurodegenerative diseases; mild cognitive impairment; humans; animals; animal models
Introduction
Disease modification remains elusive in neurodegenerative diseases (NDDs), including Alzheimer’s (AD), Parkinson’s (PD), and Huntington’s (HD) diseases. Successful interventions will be more likely made in early NDD stages, such as the mild cognitive impairment (MCI) phase of AD or, ultimately, even before clinical symptoms emerge. Pathogenic proteins,e.g., beta-amyloid (Aβ) in AD, hyperphosphorylated tau in AD and tauopathies, alpha-synuclein (αSyn) in PD, and mutant huntingtin (HTT) in HD can be present years in advance of clinically-apparent disease. Although an over-simplification, these proteins can incite microglial activation and inflammation, generating and releasing free radicals and pro-inflammatory cytokines, promoting further pathogenic protein production. If not sufficiently disposed of by the proteasome or autophagy, proteins accumulate, triggering a neurodegenerative cascade including further inflammation, proteasomal and autophagic failure, mitochondrial membrane depolarization, mitochondrial transition pore development, and mitochondrial demise, releasing free radicals and cytochrome c, in turn inducing neuronal apoptosis. Neuropsychiatric disorders, often present in the early stages of NDDs (Lauterbach, 2016), may clinically indicate psychiatric drugs (psychotropics) that simultaneously have therapeutic effects on the neurodegenerative cascade (Figures 1,2). These psychotropics are already approved by regulatory agencies. Repurposing them as NDD-modifying drugs spare regulatory and other barriers to development. This review considers existing data and the potential for six psychotropics in preventing or modifying the early course of several major NDDs.
Selected findings from neuroprotective studies of psychotropics are provided inTables 1–3, indicating a basis for considering these drugs as disease prevention or modification candidates in prodromal and early NDDs. Together, the findings suggest disease-modifying potential in AD (quetiapine, valproate, lithium, valproate plus lithium, fluoxetine, donepezil, memantine), PD (quetiapine, valproate, lithium, fluoxetine), multiple system atrophy (MSA; quetiapine, valproate), frontotemporal dementia (FTD; lithium, fluoxetine), FTD with parkinsonism linked to chromosome 17 (FTDP-17; lithium), amyotrophic lateral sclerosis (ALS; lithium, valproate plus lithium, fluoxetine),and HD (quetiapine, valproate, lithium, valproate plus lithium, fluoxetine), perhaps with differential efficacies at different NDD stages.
Preclinical animal and human clinical literature of the six most robust psychotropic attenuators of microglial activation in early NDD (Lauterbach, 2016), quetiapine, lithium, valproate, fluoxetine, donepezil, and memantine, in early-stage and pre-symptomatic AD (MCI stage), PD, and HD are reviewed here.
The literature of psychotropic trials administeredin vivoin NDD preclinical animal and clinical investigations was reviewed from the perspective of disease-modification and disease-prevention in the early and pre-symptomatic stages of NDDs recorded in the NLM PubMed database for the six drugs and three diseases of interest. Studies were limited to those judged to consider a single psychotropic commenced during pre-symptomatic or very early model pathology, excluding studies involving multiple drugs where the effects of the psychotropic of interest could not be disentangled from other drugs. Studies and models where the stage of illness was ambiguous or uncertain were not included. Tauopathies and synucleinopathies other than PD are beyond the scope of this review and also tended not to distinguish early from late stage disease. All references to MCI apply specifically to the MCI of AD. Double-blind, placebo-controlled, randomized clinical trials are designated as “DBPCRCT.”
Disease-Modification Potential in MCI of AD
Quetiapine
Although frequently prescribed in AD, quetiapine has not been studied in MCI. Quetiapine’s antidepressant and anxiolytic properties are particularly germane to MCI but somnolence, cognitive impairment, and dose-related mortality risk in advanced AD that may diminish with increasing treatment duration (Schneider et al., 2005; Piersanti et al., 2014) but is unknown in MCI might be minimized by doses lower than those required for agitation and psychosis, absent in MCI.
Lithium
In APPSwe/PS1 amyloidopathic transgenic AD mice treated with lithium (0.65 ± 0.02 mM serum level) from 10 months of age (MCI stage) for 3 months, lithium treatment was associated with decreased APP gamma cleavage, Aβ, plaques, and autophagy and improved spatial learning and memory (Zhang et al., 2011). In contrast to a PCRCT of lithium 0.5–0.8 mM with negative findings in mild AD (Hampel et al., 2009), a 12-month DBPCRCT in amnestic MCI (aMCI) involving lithium 0.25–0.5 mM (n= 45; placebon= 24) demonstrated better performance on the AD Assessment Scale-Cognitive subscale (ADAS-Cog) and attention tasks and a reduction in cerebrospinal fluid (CSF) phosphorylated tau (Forlenza et al., 2011) on lithium compared to placebo, suggesting that lowdose lithium may induce disease modification in MCI.
Micro-dose lithium 0.25 mg/kg/d in drinking water (0.006 mEq/kg, serum levels not apparent) was administered to 10-month old late MCI stage AD Cg-Tg (PDGFB-APPSwInd) amyloidopathic transgenic mice for 8 months, with lithium-treated transgenic mice demonstrating preservation of spatial and aversive stimulus recall but lithium did not prevent senile plaque development or prefrontal cortical or hippocampal dentate neuronal loss (Nunes et al., 2015). This contrasts with neuroprotection evident when lithium was administered pre-symptomatically (see below). Interestingly, in advanced AD, this same group conducted a 15-month DBPCRCT of 300 micrograms of lithium daily in 113 patients, finding a significant lack of deterioration with active treatment (–0.78 ± 0.9vs. placebo 3.37 ± 1.33), with differences becoming significant by 3 months of treatment (Nunes et al., 2013).
Single findings of low-dose lithium in human patients and micro-dose lithium in transgenic mice await replication and mechanistic discovery before undertaking larger, well-designed, disease-modifying RCTs in MCI at these dose ranges.
Valproate
In APP23 amyloidopathic transgenic AD mice, valproate begun at 7 months of age produced a four-fold reduction in neuritic plaques after 1 and 2 months treatment, partially preserving spatial memory whereas later treatment reduced plaques significantly less, indicating a disease-modifying benefit of early treatment (Qing et al., 2008). This finding was replicated in double-transgenic APP23/PS45 amyloidopathic mice at the age of 1 month (Qing et al., 2008). Plaque reduction in these investigations correlated with reductions in Aβ40and Aβ42concentrations linked to gamma-secretase inhibition, related to valproate’s inhibition of glycogen synthase kinase-3 beta (GSK-3β; Qing et al., 2008). Two weeks of valproate injections in 6 month-old APPswe/PS1dE9 amyloidopathic transgenic mice nearly completely restored contextual memory in treated mice, possibly through inhibition of histone deacetylases HDAC1, 2, 3, and/or 8 (Kilgore et al., 2010). In 5-month old Tg6799 transgenic AD mice, valproate increased hippocampal nerve growth factor (NGF) concentrations and decreased escape latencies in the Morris water maze paradigm (Noh and Seo, 2014).
These results, indicating the benefit of early treatment, suggest that the lack of slowing of cognitive, behavioral, and functional decline in the human 24-month DBPCRCT in moderately severe AD (Tariot et al., 2011) might have resulted from beginning treatment too late in the disease course, and that disease-modifying trials in MCI or pre-symptomatic AD may be warranted. In this DBPCRCT, greater hippocampal and whole brain atrophy (Fleisher et al., 2011; Tariot et al., 2011), possibly greater cognitive impairment on the Mini-Mental Status Examination (MMSE) not seen in other cognitive testing (Fleisher et al., 2011), and side-effects in the valproate group (Tariot et al., 2011) suggest the study of lower doses in future animal trials and human RCTs.
Fluoxetine
An 8-week fluoxetine DBPCRCT in 58 patients with MCI demonstrated improved MMSE and Wechsler Memory Scale III immediate and delayed logical memory scores, possibly related to hippocampal neurogenesis (Mowla et al., 2007), inviting longer term studies controlling for depression andemploying biomarkers such as hippocampal volumetry or tau and Aβ biomarkers.

Figure 1 Psychotropic effects on proteinopathy and microglial activation.
Donepezil
A 24-week DBPCRCT of donepezil 10 mg/d in 270 subjects with MCI indicated improvement of secondary efficacy measures (Salloway et al., 2004), but only improved ADAS-Cog scores on 48-week extension (Doody et al., 2009). A larger DBPCRCT in 769 aMCI patients randomized to donepezil 10 mg/d or vitamin E 2,000 I.U./d found reduced progression to AD at 12 months with donepezil but no difference in conversion from MCI to AD after 3 years, suggesting a symptomatic effect without a disease-modifying benefit (vitamin E was also without benefit) (Petersen et al., 2005). A secondary analysis of data from this study indicated that greater depression was associated with MCI conversion to AD and that donepezil reduced this conversion in this depressed group at 1.7 and 2.2 years of treatment (Lu et al., 2009). Another analysis of these data found that carriers of apolipoprotein E ε4 (APOE-ε4) and butyrylcholinesterase K* gene variants displayed an earlier age of AD onset and, under donepezil treatment, progressed from MCI to AD significantly more slowly at 3 years of treatment (De Beaumont et al., 2016). MRI data from 131 subjects in this same study revealed a non-significant trend for slowing of hippocampal atrophy rates in APOE-ε4 carriers treated with donepezil, but there were otherwise no differences between treatment groups in atrophy of the hippocampus, entorhinal cortex, whole brain, and ventricles (Jack et al., 2008). While annualized hippocampal percentage change did not differ between the groups, MRI findings in a sub-study of the 48-week DBPCRCT involving 193 MCI subjects on donepezil and 199 on placebo revealed reduced decline in total, ventricular, and cortical volumes on donepezil (Schuff et al., 2011). A French 12-month study DBPCRCT in 216 subjects with very early MCI found hippocampal volume declined 45% less (P< 0.001) in the donepezil 10 mg/d group (annualized percent change of –1.89%) less than the placebo group (–3.47%) (Dubois et al., 2015) (This study was not sufficiently powered to determine neuropsychological performance). Thus, donepezil has a mild capacity to deter the progression of MCI to AD, but the effect is too small to be advisable as a clinically – meaningful strategy except in certain subgroups.
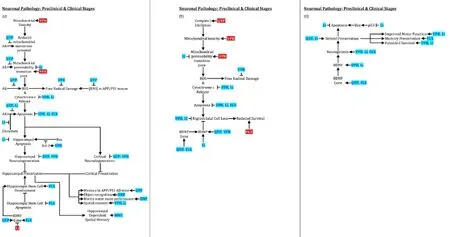
Figure 2 Psychotropic effects on neurodegeneration, neurogenesis, and behavior.
Memantine
Nine month-old 3xTg amyloidopathic/tauopathic triple transgenic AD mice in the MCI stage were treated with memantine for 3 months at human-equivalent doses, resulting in improvements in cognitive impairment and tended to reduce insoluble Aβ, Aβ dodecamers, prefibrillar soluble Aβ oligomers, fibrillar Aβ oligomers, total tau, hyperphosphorylated tau, and cognitive decline in the mild pathology group but, interestingly, achieved robust statistical significance only in advanced AD stage animals (Martinez-Coria et al., 2010). A 2-year DBPCRCT involving memantine plus galantamine in MCI was halted due to galantamine safety concerns, yielding no disease-modifying data (Peters et al., 2012).
These data suggest stage-specific windows of neuroprotective potency, with memantine disease-modification potency in moderate-to-severe AD, and less efficacy in early disease including MCI. These findings should be studied further and replicated across AD models before considering disease –modifying trials in human MCI.
Future MCI trial design considerations
Enrichment of future samples with APOE-ε4 carriers (Jack et al., 2008) and depressed subjects (Lu et al., 2009) and employing multiple year trial durations and more sensitive cognitive ratings (Fleischer et al., 2007, 2008; Salloway et al., 2008) might increase the chances of finding efficacy against MCI progression in future disease-modifying RCTs. In RCTs of drugs increasing synaptic acetylcholine levels, enrichment with depressed patients (Lu et al., 2009), APOE-ε4 carriers (Fleisher et al., 2007; Jack et al., 2008; De Beaumont et al., 2016), female butyrylcholinesterase K* carriers (De Beaumont et al., 2016), and carriers of both these genes (De Beaumont et al., 2016) may increase the odds of finding a difference from placebo. Useful biomarkers will include CSF Aβ42, tau, and phosphorylated tau and positron emission tomographic (PET) fibrillar amyloid (Albert et al., 2011), augmented by PET FDG and tau (Sperling et al., 2011), MRI medial temporal volume (DeCarli et al., 2007), and, possibly, hippocampal volumetry (Dubois et al., 2015).
Disease-Prevention Potential in Pre-Symptomatic AD
Quetiapine
Unstudied in human pre-symptomatic AD, in studies administering quetiapine beginning at 2 months in APP/PS1 mice, quetiapine decreased Aβ peptides, β-secretase expression and activity, APP C99 C-terminal fragment, cortical and hippocampal Aβ plaques, and nitrotyrosine, increased cerebral B-cell lymphoma 2 (Bcl-2), prevented memory impairment, and attenuated anxiety-like behavior at 6 and 9 months after 4 and 7 months of treatment (He et al., 2009). Findings of decreased Aβ peptides and plaques and improved memory were replicated in APP/PS1 mice treated from 4–12 months of age, in which quetiapine inhibited GSK-3β (Zhu et al., 2013). Quetiapine improved anxiety and increased cerebral BDNF at 10 months in APP/PS1 mice, suggesting a BDNF mechanism of neuroprotection (Tempier et al., 2013).Quetiapine treatment attenuated hippocampal nitrotyrosine concentrations and object recognition memory impairment at 10 months, reflecting reduced oxidative stress in APP/PS1 AD mice (Luo et al., 2014). Again in APP/PS1 mice, quetiapine decreased cortical Aβ40(but not Aβ42), almost reduced hippocampal Aβ42(P= 0.0504), decreased hippocampal microglial and cortical astroglial activation and cortical interleukin 1 beta (IL-1β) release, improved object recognition, and reduced anxiety, indicating slowing of progression, with inflammatory attenuation mediated through inhibition of the nuclear factor kappa B (NF-κB) p65 pathway and p65 nuclear translocation (Zhu et al., 2014). Thus, quetiapine administered pre-symptomatically in APP/PS1 AD transgenic mice reduced Aβ peptide species, Aβ plaques, reactive nitrosylation, impaired object recognition memory, and anxiety, possibly by inhibiting GSK-3β, glial activation, p65 nuclear translocation, and pro-inflammatory cytokines while increasing Bcl-2 and BDNF.
Lithium
Lithium prevented age-associated learning and memory impairments inDrosophilawith attenuated PS1 function (McBride et al., 2010). In P301L, 4R0N tauopathic transgenic mice, lithium decreased both tau hyperphosphorylation at AD-relevant epitopes and tau aggregation and, if started during the early stages of tangle development, lithium also reduced axonal degeneration, consistent with GSK-3 inhibition-mediated neuroprotection (Noble et al., 2005). In APP intracellular domain (AICD) transgenic AD mice associated with GSK-3β over-activation, lithium prevented tau hyperphosphorylation, tau aggregation, neurodegeneration, and working memory deficits (Ghosal et al., 2009). In the scopolamine rat model, lithium activated choline acetyltransferase, inhibited acetylcholinesterase, and, through GSK-3β inhibition, regulated dendritic spine formation and arborization, indicating improved synaptic dysfunction that develops in the pre-symptomatic phase of AD (Wu et al., 2013). Data from a transgenic Arctic mutant Aβ42adultDrosophilaAD model further indicate that Aβ42expression increases GSK-3 activity and lithium reduces Aβ42concentrations through GSK-3 inhibition (Sofola et al., 2010). In the PS1M146Lx-APPSwe-London transgenic AD mouse, lithium treatment in advance of neuropathology led to smaller Aβ plaques with reduced oligomeric halos, prevented hippocampal and entorhinal cortex neuronal loss, reduced axonal dystrophy, phosphorylated tau, autophagy, and ubiquitinated proteins, possibly related to astrocytic activation and the release of heat shock proteins (Trujillo-Estrada et al., 2013). Thus, lithium can deter incipient tauopathic, amyloidopathic, synaptopathic, and neurodegenerative progression in pre-symptomatic AD animal models through GSK-3 inhibition but these initial studies need replication.
Micro-dose lithium administered to pre-symptomatic 2-month-old Cg-Tg (PDGFB-APPSwInd) amyloidopathic AD transgenic mice at 0.25 mg/kg/d in drinking water for 16 months showed less senile plaques, an absence of hippocampal and cortical neuronal loss, evidence of increased cortical BDNF, reduced anxiety, and preserved spatial and aversive stimulus-related memory performance (Nunes et al., 2015). The authors suggest that chronic lithium treatment may inhibit calcium influx through N-methyl-D-aspartate (NMDA) receptors, increase Bcl-2, stabilize endoplasmic reticulum homeostasis, regulate autophagy by inhibiting mammalian target of rapamycin (mTOR) or activating inositol monophosphatase, or inhibit GSK-3 and extracellular regulated protein kinases (ERK) cascades. Interestingly, lithium at these doses has been considered to be incapable of inhibiting GSK-3 in humans (Tariot and Aisen, 2009), suggesting the involvement of these other mechanisms. Additional mechanisms might include attenuated neuroinflammation (Lauterbach, 2016) and mitochondrial and oxidative stress stabilization (Morris and Berk, 2016). These micro-dose findings should be replicated, mechanisms confirmed, and micro-dose lithium should then be considered in a human clinical trial in pre-symptomatic AD.
Several human epidemiologic studies indicate that lithium administration lowers the risk of AD, including a Japanese retrospective chart review of 1,423 university psychiatric clinic outpatients age 60 or more that demonstrated better MMSE scores in patients previously receiving lithiumversusage- and gender-matched controls never treated with lithium (Terao et al., 2006), a Brazilian case-control study of 66 euthymic bipolar disorder patients treated with lithium showing reduced AD prevalence relative to 48 age-, sex-, diagnosis-, and illness state-matched controls not currently so treated (Nunes et al., 2007), and a Danish study of 16,238 lithium-treated and 1,487,177 non-lithium-treated controls, revealing reduced risk of AD associated with lithium treatment (Kessing et al., 2008). These epidemiologic data collectively suggest that pre-symptomatic lithium may prevent or forestall AD in humans.
In summary, lithium ameliorated tauopathy in different animal models, reduced Aβ42inDrosophila, and can reduce synaptic dysfunction while micro-dose lithium reduced AD pathology and cognitive deficits in a transgenic mouse. Epidemiological studies found that previous lithium treatment preserved cognition (1 study) and reduced AD risk (several studies). However, a 3-week DBPCRCT of lithium (0.8 mmol serum levels) in 30 healthy subjects indicated worse performance on lithium than after discontinuation, and worse long term memory recall on lithium than in placebo (Stip et al., 2000), arguing for low- or micro-dose trials.
Valproate
In rats injected with ibotenic acid, chronic valproate increased brain histone H3 acetylation, protected nucleus basalis magnocellularis (NBM) cholinergic and gamma-aminobutyric acid-ergic (GABAergic) neurons from degeneration, and preserved cholinergic efferents to the cerebral cortex, consistent with valproate HDAC inhibition (Eleuteri et al., 2009). However, in contrast to 5-month-old Tg6799 transgenic AD mice, 1-month-old Tg6799 mice treated with valproate neither increased hippocampal NGF nor improved escape latency in the Morris maze (Noh and Seo, 2014). Further study is obviously needed to conclude any stage-dependent effects.
Fluoxetine
In aCaenorhabditis elegansmodel of AD, fluoxetine reducedAβ oligomers, delayed Aβ-induced paralysis, and extended life span, mediated through insulin signaling and the DAF-16/FOXO transcription factor (Keowkase et al., 2010). Fluoxetine prevented the loss of synaptophysin and microtubule associated protein 2, lowered brain soluble Aβ, and improved spatial memory, learning, and emotional behaviors in APP/ PS1 mice, possibly by inhibiting APP T668 phosphorylation (Wang et al., 2014). More work is needed to follow-up these initial promising findings.
Donepezil
In the Tg2576 transgenic mouse, pre-symptomatic administration of donepezil for 6 weeks did not reduce Aβ plaque progression but improved contextual and cued memory although cued memory also improved in donepezil-treated non-transgenic controls (Dong et al., 2005). A follow-up study by this group demonstrated that donepezil treatment from 3–9 months decreased brain soluble Aβ40and Aβ42and Aβ plaques and increased synaptic density in the molecular layer of the dentate gyrus in a dose-dependent manner that correlated positively with higher doses (Dong et al., 2009). In 6-week-old APP23 mice treated before pathologic development, 2 months of donepezil followed by a 3-week washout demonstrated improved Morris water maze learning and retention equivalent to non-transgenic controls, suggesting disease-modification (Van Dam et al., 2008). In APP695 V717I transgenic mice, 6 months of donepezil decreased cortical acetylcholinesterase concentrations, increased acetylcholine transferase activity and hippocampal acetylcholine concentrations, and improved spatial learning and passive avoidance test memory (Cai et al., 2011). Again in APPV717I mice, donepezil administered pre-symptomatically for 4 months decreased cortical GSK-3β expression, suggesting a potential mechanism of donepezil (Shi et al., 2013). In APPswe/PS1dE9, donepezil administered pre-symptomatically for 6 months until 10.5 months of age revealed decreased Aβ concentrations, plaques, and memory impairment (Jeon et al., 2011). In rats receiving intra-cisternal Aβ42, concomitant donepezil decreased amyloid deposition, prevented cholinergic dysfunction, and improved spatial and avoidance memory (Wu et al., 2014).
A 2-week DBPCRCT of donepezil 5 mg/d in 27 healthy adults showed no differences between groups on neuropsychological testing although the small sample size, low-dose, and minimal duration of treatment impede interpretation (Beglinger et al., 2004). Although long-term cognitive outcomes were not determined, an 8-week PCRCT of donepezil 5–10 mg/d in healthy men in their 7thdecade of life revealed a 31% increase in insulin-like growth hormone and a doubling of growth hormone, reversing age-associated declines and suggesting possible mechanisms by which donepezil might reduce AD progression (Obermayr et al., 2005).
Pre-symptomatic donepezil treatment in AD animal model has demonstrated decreased Aβ and Aβ plaques and improved acetylcholine concentrations and memory outcomes across different models, but no adequate trials in humans have yet been conducted.
Memantine
In Wistar rats pretreated for 3 days with memantine before receiving intra-cerebroventricular Aβ42oligomer injections into the right NBM and frontal cortex, followed by memantine treatment for another 7 days until sacrifice, memantine attenuated microglial activation, preserved NBM cholinergic efferents to the neocortex, and decreased attention and memory impairments (Nyakas et al., 2011). APP23 mice treated pre-symptomatically for 2 months followed by a 3-week washout period dose-dependently improved learning and spatial recall consistent with disease modification (Van Dam and De Deyn, 2006). Memantine treatment for 6 weeks initiated 24 hours before Aβ and ibotenate injections to the bilateral hippocampi reduced microglial activation and hippocampal neuronal damage and prevented learning deficits in a dose-dependent manner (Nakamura et al., 2006). In rats undergoing intracerebroventricular Aβ25–35, memantine prevented microglial and astrocytic activation and attenuated peptidylarginine deiminase 2 (Arif and Kato, 2009). Tg2576 mice treated with memantine for 6 months showed reduced Aβ plaque deposition and increased hippocampal and cortical synaptic density at 5 mg/kg yet without improvement in a fear conditioning paradigm, while 10–20 mg/kg doses had the same effects but were associated with increased neuronal degeneration (Dong et al., 2008). In APP/PS1 mice treated from ages 3–7 months, memantine was associated with reduced Aβ plaque burden and prevention of object recognition memory impairment (Scholtzova et al., 2008). In the 6-month old 3xTg mouse, 3 months of memantine reduced insoluble Aβ, Aβ dodecamers, prefibrillar soluble oligomers, fibrillar oligomers, total tau, hyperphosphorylated tau, and cognitive decline (Martinez-Coria et al., 2010). In Wistar rats treated with intrahippocampal okadaic acid and pretreated with memantine, CSF glutamate levels and spatial memory impairment were decreased compared to okadaic acid-exposed controls not treated with memantine, possibly by decreasing glutamate, tau hyperphosphorylation, and Cdk5/ p25 signaling (Zimmer et al., 2012).
Future pre-symptomatic AD trial design considerations
Enhanced risk of progression from pre-symptomatic AD to MCI may be increased by enriching samples with patients with first-onset of anxiety, a predictor of incident cognitive impairment with a relative risk of 1.77, 95%CI1.38–2.26 (Gulpers et al., 2016). Newer neuropsychological instruments are under development and may be more sensitive in detecting progression to MCI, such as the Loewenstein– Acevedo Scale for Semantic Interference and Learning (Loewenstein et al., 2016). Useful biomarkers in monitoring progression from the pre-symptomatic to MCI stage will include CSF Aβ42, and PET amyloid and temporoparietal glucose metabolism (Sperling et al., 2011), augmented by CSF or PET tau, which progresses in this phase (Sperling et al., 2011) and, possibly, hippocampal/medial temporal volumetry (Dubois et al., 2015) and translocator protein (TSPO) receptor ligand PET (Lauterbach, 2016).
Disease-Modification Potential in Early PD
Studies of disease modification in early PD are limited to animal models for lithium and valproate, and human disease-modifying trials were not apparent for any of the psychotropics.
曾晓东:对程校长来说,他不仅是一个领导,还是一个老大哥,带这么一支年轻的教师队伍,对他的智力和耐心肯定都是一个很大的挑战。
Quetiapine
Quetiapine doubled the 180-day mortality rate (hazard ratio of death 2.16, 95% confidence interval 1.88–2.48) in PD (Weintraub et al., 2016) and, if this risk also applies in early stage PD, lower doses than those applied in PD psychosis and agitation might avoid this concern, slow PD progression, and extend survival.
Lithium
In vitroevidence suggests that lithium enhances autophagy by inhibiting myo-inositol-1,4,5-trisphosphate (MITP) in PD (Motoi et al., 2014). A study of lithium carbonate given for 7 days after 7-day 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) administration revealed increased autophagy, substantia nigra dopamine neurons, and levels of striatal dopamine and 3,4-dihydroxyphenylacetic acid (DOPAC) and produced improvements in rolling bar latency, pole-climbing time, and spontaneous motor activity, perhaps related to increased autophagy (Li et al., 2013). These findings await replication but suggest disease-modification after early introduction of lithium in this model.
Valproate
Valproate was co-administered with MPTP for 5 days and 14 days afterwards to male FVBn mice throughout the period of active neurodegeneration, partially preventing striatal dopamine depletion with nearly complete protection of the substantia nigra against dopaminergic cell loss, possibly by HDAC inhibition (Kidd and Schneider, 2011). A study of valproate for 7 days following MPTP administration demonstrated increased autophagy, higher levels of substantia nigra dopamine neurons and striatal dopamine, and improved rolling bar latency, pole-climbing time, and spontaneous activity, possibly due to autophagic/lysosomal activation (Li et al., 2013). Valproate in rats undergoing unilateral striatal injections with 6-hydroxydopamine decreased HDAC, microcytic and astrocytic activation, tumor necrotic factor-alpha, striatal and dopaminergic mesencephalonic neuronal loss, and behavioral impairments, related to anti-inflammatory activity (Ximenes et al., 2015). The proteasomal inhibitor lactacystin was injected into the substantia nigra of rats in a model of PD and, 7 days later, valproate 200 mg/kg or 400 mg/kg daily was given for 28 days, dose-dependently reducing midbrain neurodegeneration and improving motor deficits, possibly by reversing histone hypoacetylation, and inducing neuroprotective and neurotrophic factors (Harrison et al., 2015). In this same intranigral lactacystin model under the same dosing conditions, valproate dose-dependently protected substantia nigra dopamine neurons and prevented spreading of neuronal degeneration to ventral tegmental area dopamine neurons (Harrison et al., 2016).
Future early PD trial design considerations
Enrichment of samples with elderly, men, occupational exposure to pesticides or solvents, life-long abstainers from caffeine and tea and smoking, with a history of REM behavior disorder (RBD), mild PD signs, olfactory loss, constipation, excessive daytime somnolence, hypotension, erectile dysfunction, urinary dysfunction, or depression, with siblings with early onset PD or first-degree relatives with PD will increase the likelihood of PD progression (Berg et al., 2015). The use of prodromal PD criteria (Berg et al., 2015) can assist diagnostic validity and sample homogeneity. Biomarkers might include fluoro-DOPA PET or dopamine transporter SPECT imaging and, perhaps, substantia nigra hyperechogenicity.
Disease-Prevention Potential in Pre-Symptomatic PD
No data were found for quetiapine, donepezil, or memantine in pre-symptomatic PD.
Lithium
Pretreatment of MPTP mice with lithium 4.4 g/kg (3.3 g/kg produced human therapeutic serum levels) for 4 weeks revealed dose-dependent protection against the depletion of striatal dopamine, DOPA, homovanillic acid, and Bcl-2 while decreasing apoptotic Bax, suggesting lithium’s ability to prevent neuronal apoptosis (Youdim and Arraf, 2004). In the intranasal MPTP rat, lithium pretreatment for seven days prevented the loss of striatal dopamine and shortterm memory impairment (Castro et al., 2012). Low-dose lithium (0.2 mEq/L blood level) prevented striatal microglial activation, astrogliosis, and further dopaminergic degeneration while preventing motor impairment in the aged parkin transgenic PD mouse model, possibly by anti-inflammatory effects (Lieu et al., 2014). In the A53T mouse model of PD also exposed to paraquat and maneb, lithium at human therapeutic concentrations prevented accumulation of oxidized/nitrated αSyn in the substantia nigra and striatum and oxidative stress-induced neurodegeneration (Kim et al., 2011). In the 6-OHDA rat, lithium did not reduce substantia nigra pars compacta dopaminergic degeneration despite inhibiting GSK-3β (Yong et al., 2011). These data, indicating prevention of nigrostriatal degeneration in different PD models, now await replication at specific doses.
Valproate
Nuclear αSyn prevents histone acetylation by histone acetyltransferase (Kontopoulos et al., 2006), possibly reversedby valproate-inhibition of HDACs. Valproate protected dopaminergic neurons inCaenorhabditis elegansover-expressing αSyn, attributed to ERK-MAPK inhibition (Kautu et al., 2013). Chronic valproate decreased nigral and striatal monoubiquitinated αSyn nuclear translocation and preserved substantia nigral neurons, nigral and striatal tyrosine hydroxylase, and striatal dopamine levels in the rotenone rat PD model, presumably by valproate HDAC inhibition, increasing histone H3 acetylation (Monti et al., 2010). In another rotenone rat study, chronic valproateprevented loss of nigral dopamine neurons, nigral and striatal tyrosine hydroxylase, and contralateral forelimb disuse (Carriere et al., 2014). In the intranasal MPTP rat, seven days pretreatment with valproate prevented striatal dopamine loss (Castro et al., 2012). Replication of these pre-lesion valproate studies will be desirable across animal models of PD.
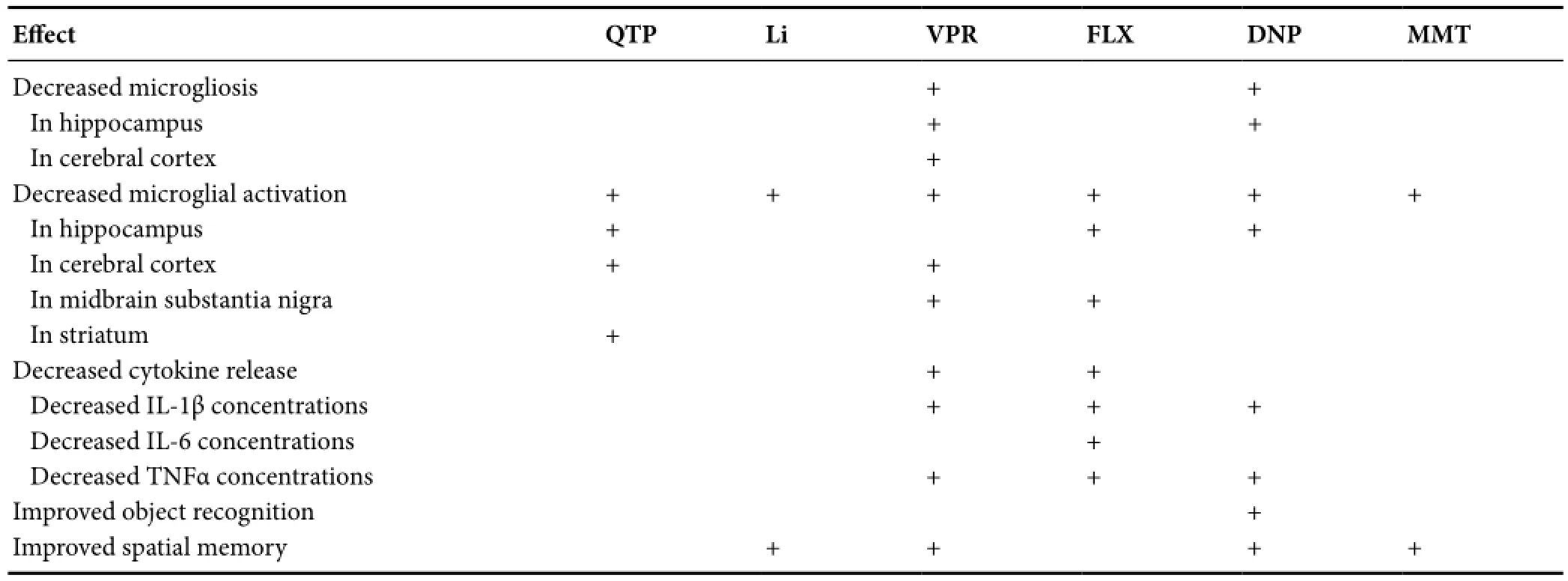
Table 1 Psychotropic effects on microgliosis, microglial activation, cytokine release, and associated cognitive performance
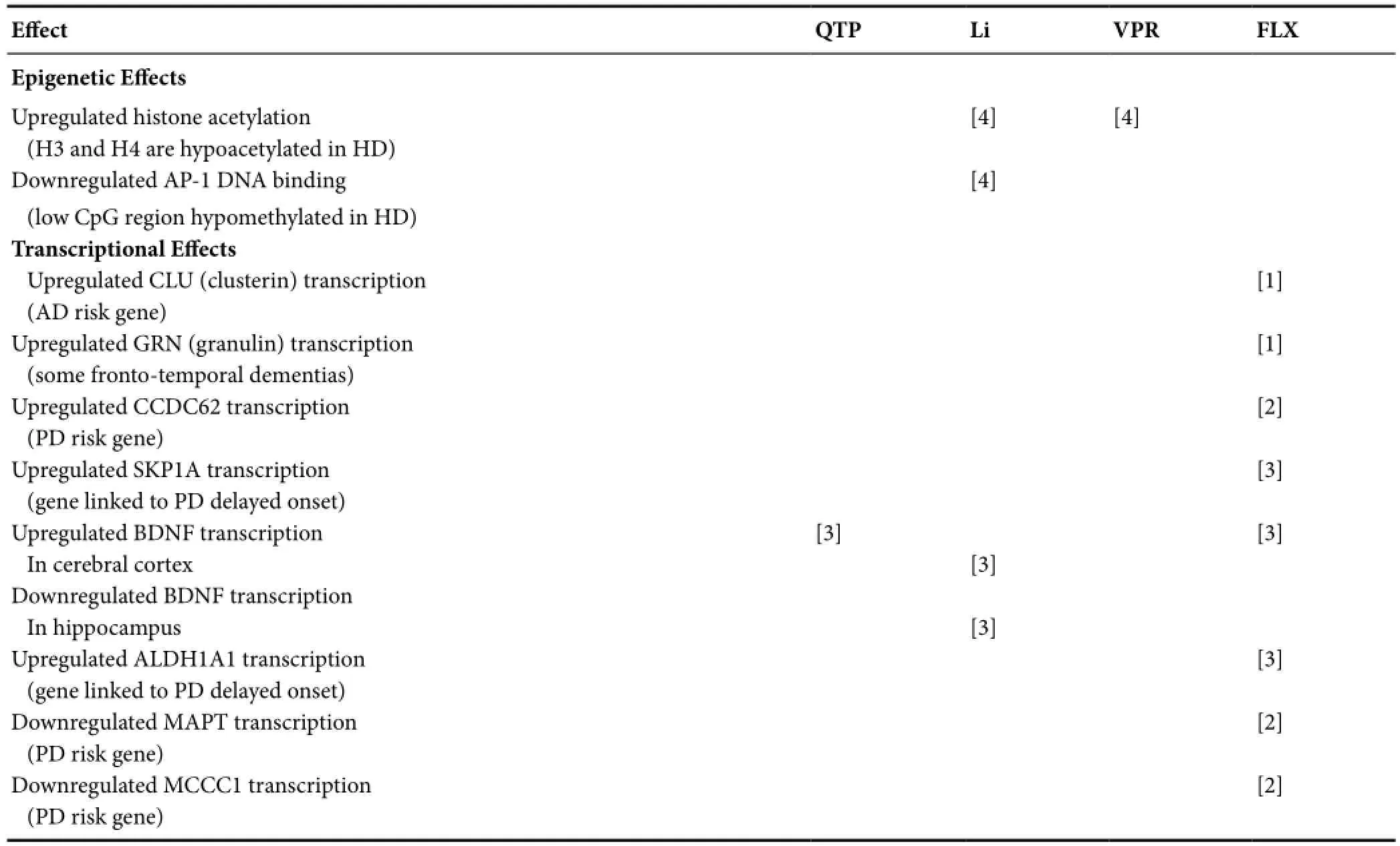
Table 2 Epigenetic and transcriptional effects of psychotropics
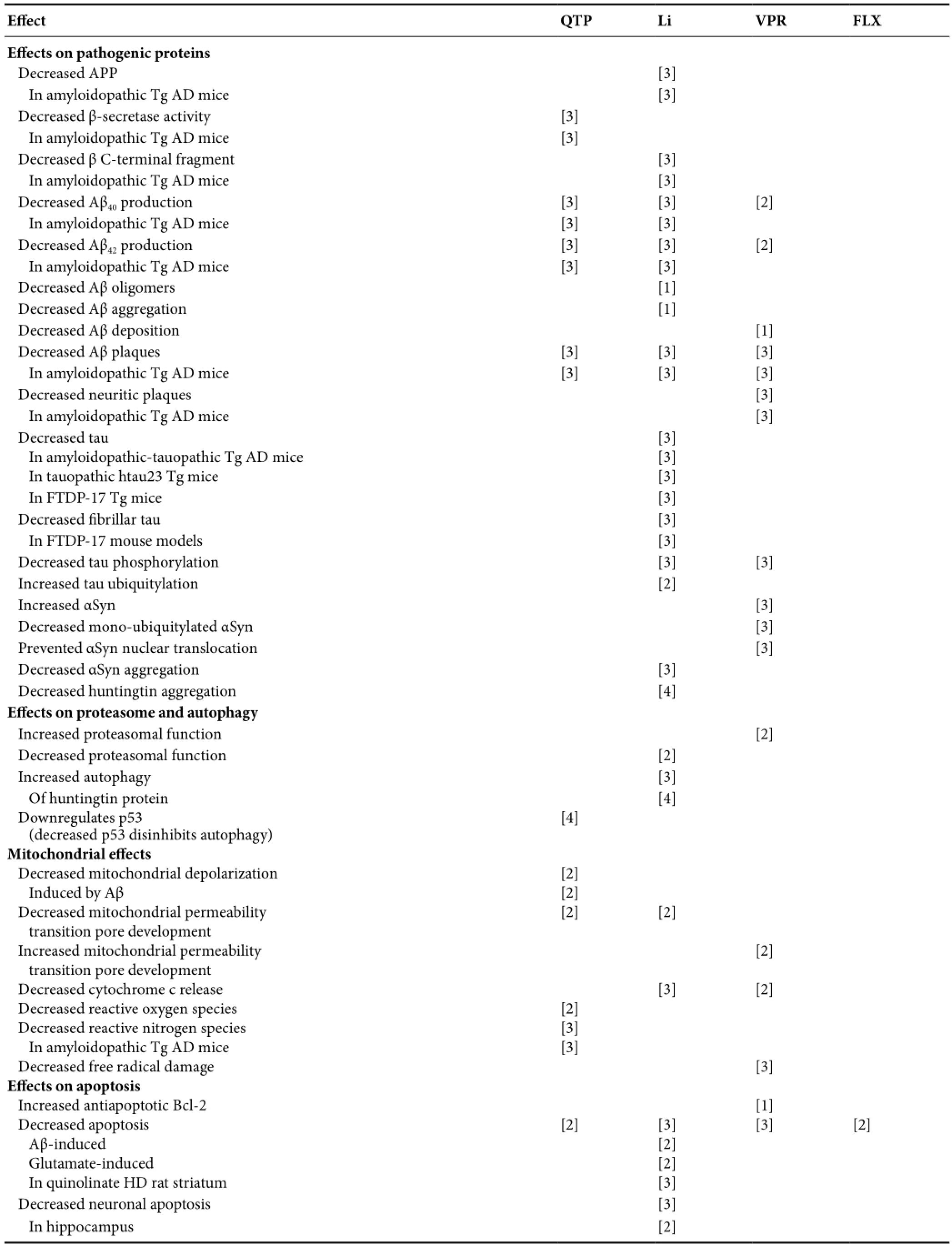
Table 3 Replicated psychotropic effects on protein, proteasome, autophagy, mitochondria, apoptosis, neurodegeneration, neurogenesis, and animal cognition and behavior
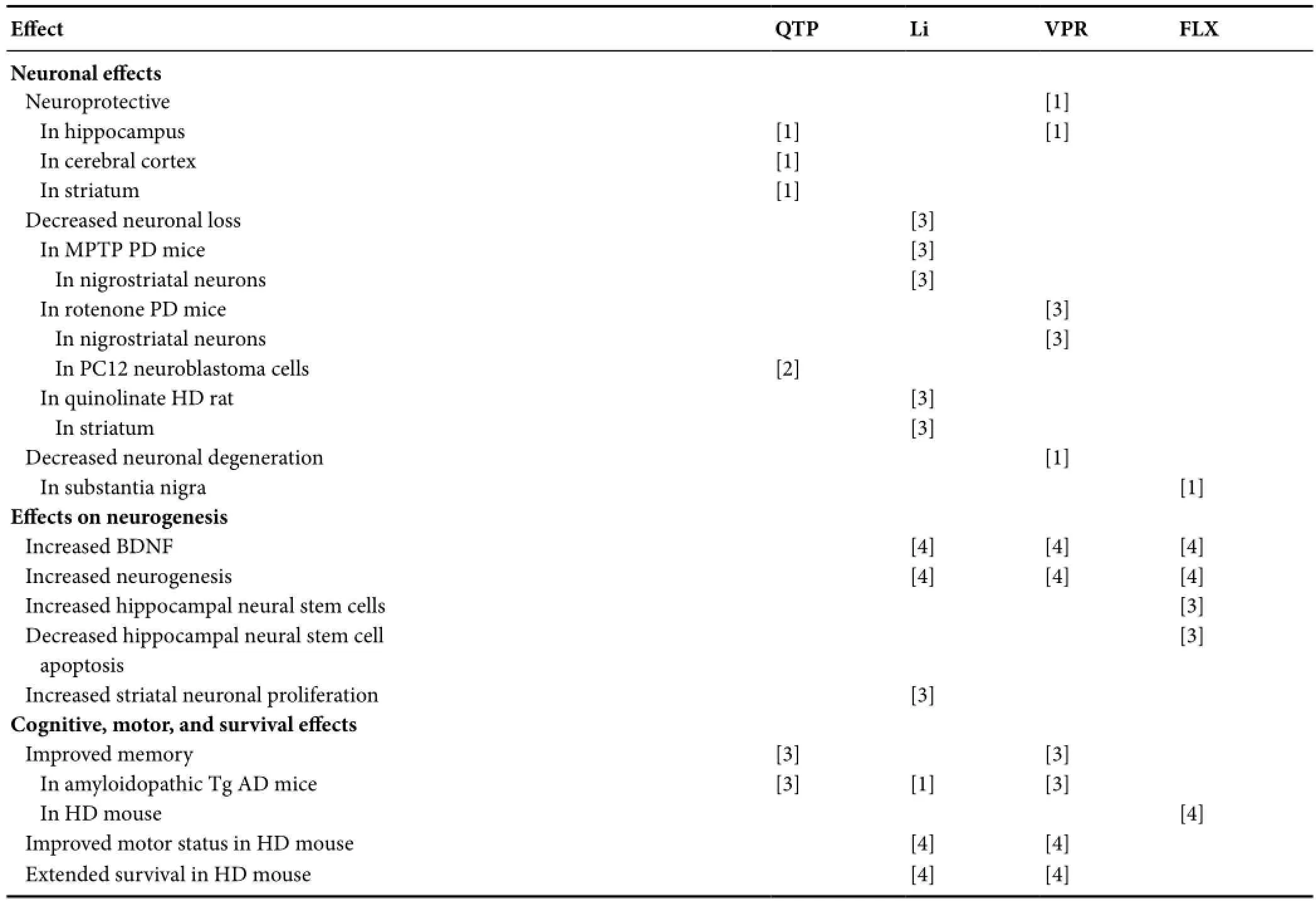
Table 3 Continued
Fluoxetine
In the MPTP mouse, fluoxetine injections beginning when nigrostriatal dopamine neurons had been depleted by 15% prevented further degenerative depletion and increased striatal dopamine levels producing partial motor recovery, related to attenuated microglial activation (Chung et al., 2011). In 4–5 month-old transgenic A53T αSyn mice, chronic fluoxetine reversed hippocampal dysfunction and neurogenesis impairment, increasing hippocampal dentate gyrus neural precursor cell proliferation and differentiation into neurons, ascribed to increased BDNF and GDNF levels (Kohl et al., 2012). These findings await replication.
Future pre-symptomatic PD trial design considerations
Sample enrichment with subjects having reduced dopaminergic markers on functional imaging, substantia nigra hyperechogenicity, hyposmia, constipation, history of RBD (Mahlknecht et al., 2015), and other features listed in“Future early PD trial design considerations” may enhance the probability of PD progression. Biomarkers include dopaminergic functional imaging and, possibly, tegmental diffusion tensor imaging, supplementary motor area functional magnetic resonance imaging, and dorsolateral substantia nigra T2-weighted 7 Tesla MRI (Mahlknecht et al., 2015).
Disease-Modification in Early HD
There were no data relevant to disease modification in early HD for quetiapine or valproate.
Lithium
R6/2 HD transgenic mice treated with lithium demonstrated improved rotarod motor performance but not prolongedsurvival (Wood and Morton, 2003).
Several early DBPC studies of lithium in 6 HD patients each showed no symptomatic benefit for chorea over 3–6 weeks but did not observe for HD progression (Leonard et al., 1975; Vestergaard et al., 1977). A clinical case series of 3 patients documented HD non-progression over 3–4 years with low-dose lithium 150–300 mg/d along with carbamazepine (Danivas et al., 2013). These findings suggest a disease-modifying effect without symptomatic benefit.
Fluoxetine
A 4-month DBPCRCT of fluoxetine in 30 non-depressed HD patients found no difference in neurological, cognitive, behavioral, or functional outcomes in this small study of brief duration (Como et al., 1997). Two HD patients treated with fluoxetine experienced non-progression over the subsequent 2 and 6 years, respectively (De Marchi et al., 2001). These findings suggest a disease-modifying effect without symptomatic benefit.
Donepezil
A 12-week DBPCRCT in 30 adults with HD lacking depression and dementia, with other medications held constant during the trial, showed no improvement in chorea, cognition, or quality of life with donepezil at 12 weeks, although chorea, symbol digit, ADAS-Cog, and WAIS-III symbol searching did not progress, in contrast to placebo group deteriorations (Cubo et al., 2006), suggesting disease-modification potentially detectable in a larger and longer trial.
Memantine
A patient with HD experienced non-progression over 6 months after adding memantine to the regimen (Cankurtaran et al., 2006). A 3-month un-blinded memantine trial in 12 patients showed improvement only in chorea but did not observe for disease-modification (Ondo et al., 2007). An un-blinded memantine trial in 4 patients showed neuropsychological non-progression at 18 months in one patient whereas the other 3 patients, who had discontinued memantine after 3–4 months, showed minor progression at 12 months (Hjermind et al., 2011). A two-year, un-blinded, flexible-dose trial of memantine in 27 patients suggested slowing of HD progression relative to historical controls (Beister et al., 2004). Blinded controlled clinical trials of suitable durations are now needed.
Future early (prodromal) HD trial design considerationsPre-specified criteria (Reilmann et al., 2014) and HD gene repeat number may enhance sample homogeneity. Subjects can be followed for progression to end-points that may include motor, cognitive, and striatal volume outcomes.
Disease-Prevention in Pre-Symptomatic HD
There were no data for quetiapine or donepezil. Preliminary HD animal model data support the notion that lithium may prevent, valproate may delay, and memantine may slow HD emergence during the pre-symptomatic phase but replications are needed.
Lithium
Lithium protected against optic neurodegeneration in an N171-120QDrosophilatransgenic HD model (Sarkar et al., 2008). Lithium dose-dependently increased neuronal survival in N171-150QCaenorhabditis elegans(Voisine et al., 2007). Lithium pretreatment increased Bcl-2 and reduced striatal neuronal apoptosis and lesion volume in the HD quinolinate rat model (Wei et al., 2001). In the 3-nitropropionic acid (3-NPA) HD rat, lithium prevented oxidative stress, cellular pathology, motor impairments, and weight loss, potentiated by hemin, with this synergy reversed by protoporphyrin, suggesting hemeoxygenase-1 and GSK-3β involvement in HD pathophysiology (Khan et al., 2015).In vitroevidence suggests lithium may enhance autophagy in HD by inhibiting MITP (Motoi et al., 2014).
Pre-symptomatic N171-82Q transgenic HD mice fed lithium at human therapeutic levels showed increased cortical GSK-3β inhibition, cortical and striatal HDAC inhibition, cortical and striatal BDNF increases, and reduced behavioral depression but neurological status, motor skill learning, and lifespan did not improve, in contrast to synergistic beneficial effects with combined lithium and valproate treatment (Chiu et al., 2011). The same findings obtained in similarly-treated YAC128 mice although neurological status and anxiety did not improve, in contrast to synergistic beneficial effects with combined lithium and valproate treatment (Chiu et al., 2011).
Pre-symptomatic micro-dose lithium in YAC128 transgenic HD mice normalized caspase-6 activation and BDNF levels, prevented loss of striatal neurons and volume, and improved neurological dysfunction (Pouladi et al., 2012).
Valproate
In N171-82Q transgenic HD mice, high dose valproate improved motor activity and survival, delaying symptom onset by 31% (Zádori et al., 2009). In another N171-82Q mouse study, dietary valproate producing human therapeutic serum levels inhibited cortical GSK-3β and cortical and striatal HDAC, increased cortical and striatal BDNF, the molecular chaperone HSP70, and lifespan, and improved behavioral depression but did not improve neurological status or motor skill learning, in contrast to synergistic beneficial effects in mice treated with combined lithium and valproate treatment (Chiu et al., 2011). In similarly-treated YAC128 mice, valproate produced the same positive effects but did not improve neurological status or anxiety, in contrast to synergistic beneficial effects of combined lithium and valproate treatment (Chiu et al., 2011).
Fluoxetine
In R6/1 HD transgenic mice, fluoxetine increased hippocampal neurogenesis, preserved dentate gyrus volume, preserved spatial memory, and improved depressive behavior (Grote et al., 2005).
Memantine
Memantine co-administered with 3-NPA in rats resulted in partially preserved striatal cells and striatal volume, increased Bcl-xl concentrations, and decreased huntingtin proteolytic fragments, Bax levels, apoptosis, degenerating neurons, weight loss, and micro-calpain levels (Lee et al., 2006), suggesting memantine attenuates NMDA receptor-related calcium-mediated apoptosis in this model.
Future pre-symptomatic HD trial design considerations
Future human trials should ideally involve blinding, placebo-treated controls, and adequate duration in disease-modifying or preventive designs. Pre-symptomatic trials can enlist family members with known genotype and CAG repeat number to approximate time of disease onset. Pre-specified criteria,e.g., Reilmann et al. (2014), may enhance sample selection and homogeneity. Progression to motor, cognitive, and striatal volume end-points might increase signal detection.
Summary
Failed clinical trials in advanced NDDs should not deter consideration of the above psychotropics for trials in pre-symptomatic and early NDDs since pathophysiologies and drug effects vary between disease stages and therapeutic failure in late disease does not necessarily mean failure in early disease.
In MCI, clinical trials suggest disease-modifying potential for low-dose lithium and, in selected patients, donepezil, findings that await replication, while replicated transgenic animal data encourage further study of valproate in AD MCI, especially at subclinical doses; quetiapine awaits study while fluoxetine animal and memantine human studies are presently inconclusive; a memantine animal study suggested less disease-modifying potency in MCI than in later AD and should be replicated before considering further human trials.In pre-symptomatic AD, quetiapine has consistently reduced Aβ peptides, Aβ plaques, reactive nitrogen species, and improved object recognition memory and anxiety in APP/PS1 mice. Epidemiological data indicate lithium reduces human AD risk and, in animal models, ameliorates tauopathy and improves cognition but several amyloidopathic benefits await replication, as do animal findings for valproate and fluoxetine. In animal models, donepezil treatment has decreased Aβ and plaques, increased acetylcholine levels, and improved memory, awaiting exploration in humans. Memantine has consistently reduced microglial activation, Aβ plaques, hippocampal degeneration, and memory impairment in several animal models.
In early PD, valproate reduced substantia nigra degeneration and improved motor performance in animals. Other drugs await study and preclinical lithium findings await replication.In pre-symptomatic PD, lithium and valproate each prevented both substantia nigra degeneration and striatal dopamine depletion in animals. Additional findings for lithium, valproate, and fluoxetine await replication and human studies are needed. Donepezil and memantine await investigation.
In early HD, uncontrolled clinical data indicate non-progression with lithium, fluoxetine, donepezil, and, especially, memantine, urging blinded controlled clinical trials. Animal data are limited to a single (positive) study of lithium.In pre-symptomatic HD, animal studies of lithium at varying doses indicate preserved striatal volume and neurological function but other positive findings for lithium, valproate, fluoxetine, and memantine await replication. Clinical data in humans are needed for all six drugs.
Conclusion
Substantial data suggest promising potential for these psychotropics as disease-modifiers in pre-symptomatic and early-stage NDDs. Preclinical findings should be replicated across a variety of disease-specific animal models, especially at subclinical doses, subsequently confirmed in blinded, controlled clinical trials employing appropriate disease-modifying designs (delayed-start, randomized withdrawal,etc.) and methodological refinements detailed above for each disease and stage. Microglial activation, important in early NDDs, might additionally be tracked by serial PET quantitation using TSPO receptor ligands (Lauterbach, 2016). The findings summarized here offer the exciting prospect of minimizing NDD progression and, ultimately, the potential for arresting these diseases before signs and symptoms even begin.
Acknowledgments:The author thanks John T. Knight, B.F.A., Photography and Graphic Arts Department, Mercer University School of Medicine, USA for his assistance in developing the Figures.
Author contributions:As the sole author, the author was entirely and exclusively involved in the conception, design, data collection, analysis, and interpretation of the data and revision, technical support (except for figure production, as indicated under Acknowledgments), and writing of the manuscript (there were no funding, administrative, material, or supervisory functions involved).
Conflicts of interest:
· Shareholder, Pfizer, Inc., < $3600, 1999 – present;
· Speaker, “Understanding The Relationship Of Pseudobulbar Affect (PBA) & Traumatic Brain Injury (TBI), 2015, PsychU website, managed by OPEN MINDS;
· Consultant and Speaker, Otsuka America Pharmaceutical, Inc., 2015-2016; · Discussant/Speaker, “A Discussion Of Pseudobulbar Affect,” Medscape (Division of WebMD), 2016.
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH (2011) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging–Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7:270-279.
Arif M, Kato T (2009) Increased expression of PAD2 after repeated intracerebroventricular infusions of soluble Abeta(25-35) in the Alzheimer’s disease model rat brain: effect of memantine. Cell Mol Biol Lett 14:703-714.
Beglinger LJ, Gaydos BL, Kareken DA, Tangphao-Daniels O, Siemers ER, Mohs RC (2004) Neuropsychological test performance in healthy volunteers before and after donepezil administration. J Psychopharmacol 18:102-108.
Beister A, Kraus P, Kuhn W, Dose M, Weindl A, Gerlach M (2004) The N-methyl-D-aspartate antagonist memantine retards progression of Huntington’s disease. J Neural Transm Suppl 68:117-122.
Berg D, Postuma RB, Adler CH, Bloem BR, Chan P, Dubois B, Gasser T, Goetz CG, Halliday G, Joseph L, Lang AE, Liepelt-Scarfone I, Litvan I, Marek K, Obeso J, Oertel W, Olanow CW, Poewe W, Stern M, Deuschl G (2015) MDS research criteria for prodromal Parkinson’s disease. Mov Disord 30:1600-1609.
Cai LL, Li H, Liu JG, Liu LT, Guan J, Liu MF, Hu J, Wei Y (2011) Effects of early intervention with Huannao Yicong formula effective components on behavior and cholinergic system of β-amyloid precursor protein transgenic mice. Zhong Xi Yi Jie He Xue Bao 9:292-298.
Cankurtaran ES, Ozalp E, Soygur H, Cakir A (2006) Clinical experience with risperidone and memantine in the treatment of Huntington’s disease. J Natl Med Assoc 98:1353-1355.
Carriere CH, Kang NH, Niles LP (2014) Neuroprotection by valproic acid in an intrastriatal rotenone model of Parkinson’s disease. Neuroscience 267:114-121.
Castro AA, Ghisoni K, Latini A, Quevedo J, Tasca CI, Prediger RD (2012) Lithium and valproate prevent olfactory discrimination and short-term memory impairments in the intranasal 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) rat model of Parkinson’s disease. Behav Brain Res 229:208-215.
Chiu CT, Liu G, Leeds P, Chuang DM (2011) Combined treatment with the mood stabilizers lithium and valproate produces multiple beneficial effects in transgenic mouse models of Huntington’s disease. Neuropsychopharmacology 36:2406-2421.
Chung YC, Kim SR, Park JY, Chung ES, Park KW, Won SY, Bok E, Jin M, Park ES, Yoon SH, Ko HW, Kim YS, Jin BK (2011) Fluoxetine prevents MPTP-induced loss of dopaminergic neurons by inhibiting microglial activation. Neuropharmacology 60:963-974.
Como PG, Rubin AJ, O’Brien CF, Lawler K, Hickey C, Rubin AE, Henderson R, McDermott MP, McDermott M, Steinberg K, Shoulson I (1997) A controlled trial of fluoxetine in nondepressed patients with Huntington’s disease. Mov Disord 12:397-401.
Cubo E, Shannon KM, Tracy D, Jaglin JA, Bernard BA, Wuu J, Leurgans SE (2006) Effect of donepezil on motor and cognitive function in Huntington disease. Neurology 67:1268-1271.
Danivas V, Moily NS, Thimmaiah R, Muralidharan K, Purushotham M, Muthane U, Jain S (2013) Off label use of lithium in the treatment of Huntington’s disease: A case series. Indian J Psychiatry 55:81-83.
De Beaumont L, Pelleieux S, Lamarre-Théroux L, Dea D, Poirier J; and the Alzheimer’s Disease Cooperative Study (2016) Butyrylcholinesterase K and apolipoprotein E-e4 reduce the age of onset of Alzheimer’s disease, accelerate cognitive decline, and modulate donepezil response in mild cognitively impaired subjects. J Alzheimers Dis 54:913-922.
De Marchi N, Daniele F, Ragone MA (2001) Fluoxetine in the treatment of Huntington’s disease. Psychopharmacology (Berl) 153:264-266.
DeCarli C, Frisoni GB, Clark CM, Harvey D, Grundman M, Petersen RC, Thal LJ, Jin S, Jack CR Jr, Scheltens P; Alzheimer’s Disease Cooperative Study Group (2007) Qualitative estimates of medial temporal atrophy as a predictor of progression from mild cognitive impairment to dementia. Arch Neurol 64:108-115.
Dong H, Yuede CM, Coughlan CA, Murphy KM, Csernansky JG (2009) Effects of donepezil on amyloid-beta and synapse density in the Tg2576 mouse model of Alzheimer’s disease. Brain Res 1303:169-178.
Dong H, Yuede CM, Coughlan C, Lewis B, Csernansky JG (2008) Effects of memantine on neuronal structure and conditioned fear in the Tg2576 mouse model of Alzheimer’s disease. Neuropsychopharmacology 33:3226-3236.
Dong H, Csernansky CA, Martin MV, Bertchume A, Vallera D, Csernansky JG (2005) Acetylcholinesterase inhibitors ameliorate behavioral deficits in the Tg2576 mouse model of Alzheimer’s disease. Psychopharmacology (Berl) 181:145-152.
Doody RS, Ferris SH, Salloway S, Sun Y, Goldman R, Watkins WE, Xu Y, Murthy AK (2009) Donepezil treatment of patients with MCI: a 48-week randomized, placebo-controlled trial. Neurology 72:1555-1561.
Dubois B, Chupin M, Hampel H, Lista S, Cavedo E, Croisile B, Louis Tisserand G, Touchon J, Bonafe A, Ousset PJ, Ait Ameur A, Rouaud O, Ricolfi F, Vighetto A, Pasquier F, Delmaire C, Ceccaldi M, Girard N, Dufouil C, Lehericy S, et al. (2015) Donepezil decreases annual rate of hippocampal atrophy in suspected prodromal Alzheimer’s disease. Alzheimers Dement 11:1041-1049.
Eleuteri S, Monti B, Brignani S, Contestabile A (2009) Chronic dietary administration of valproic acid protects neurons of the rat nucleus basalis magnocellularis from ibotenic acid neurotoxicity. Neurotox Res 15:127-132.
Fleisher AS, Truran D, Mai JT, Langbaum JB, Aisen PS, Cummings JL, Jack CR Jr, Weiner MW, Thomas RG, Schneider LS, Tariot PN; Alzheimer’s Disease Cooperative Study (2011) Chronic divalproex sodium use and brain atrophy in Alzheimer disease. Neurology 77:1263-1271.
Fleisher AS, Sun S, Taylor C, Ward CP, Gamst AC, Petersen RC, Jack CR Jr, Aisen PS, Thal LJ (2008) Volumetric MRI vs clinical predictors of Alzheimer disease in mild cognitive impairment. Neurology 70:191-199.
Fleisher AS, Sowell BB, Taylor C, Gamst AC, Petersen RC, Thal LJ; Alzheimer’s Disease Cooperative Study (2007) Clinical predictors of progression to Alzheimer disease in amnestic mild cognitive impairment. Neurology 68:1588-1595.
Forlenza OV, Diniz BS, Radanovic M, Santos FS, Talib LL, Gattaz WF (2011) Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry 198:351-356.
Ghosal K, Vogt DL, Liang M, Shen Y, Lamb BT, Pimplikar SW (2009) Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc Natl Acad Sci U S A 106:18367-18372.
Grote HE, Bull ND, Howard ML, van Dellen A, Blakemore C, Bartlett PF, Hannan AJ (2005) Cognitive disorders and neurogenesis deficits in Huntington’s disease mice are rescued by fluoxetine. Eur J Neurosci 22:2081-2088.
Gulpers B, Ramakers I, Hamel R, Köhler S, Oude Voshaar R, Verhey F (2016) Anxiety as a predictor for cognitive decline and dementia: a systematic review and meta-analysis. Am J Geriatr Psychiatry 24:823-842.
Hampel H, Ewers M, Bürger K, Annas P, Mörtberg A, Bogstedt A, Frölich L, Schröder J, Schönknecht P, Riepe MW, Kraft I, Gasser T, Leyhe T, Möller HJ, Kurz A, Basun H (2009) Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry 70:922-931.
Harrison IF, Anis HK, Dexter DT (2016) Associated degeneration of ventral tegmental area dopaminergic neurons in the rat nigrostriatal lactacystin model of parkinsonism and their neuroprotection by valproate. Neurosci Lett 614:16-23.
Harrison IF, Crum WR, Vernon AC, Dexter DT (2015) Neurorestoration induced by the HDAC inhibitor sodium valproate in the lactacystin model of Parkinson’s is associated with histone acetylation and up-regulation of neurotrophic factors. Br J Pharmacol 172:4200-4215.
He J, Luo H, Yan B, Yu Y, Wang H, Wei Z, Zhang Y, Xu H, Tempier A, Li X, Li XM (2009) Beneficial effects of quetiapine in a transgenic mouse model of Alzheimer’s disease. Neurobiol Aging 30:1205-1216.
Hjermind LE, Law I, Jønch A, Stokholm J, Nielsen JE (2011) Huntington’s disease: effect of memantine on FDG-PET brain metabolism? J Neuropsychiatry Clin Neurosci 23:206-210.
Jack CR Jr, Petersen RC, Grundman M, Jin S, Gamst A, Ward CP, Sencakova D, Doody RS, Thal LJ; Members of the Alzheimer’s Disease Cooperative Study (ADCS) (2008) Longitudinal MRI findings from the vitamin E and donepezil treatment study for MCI. Neurobiol Aging 29:1285-1295.
Jeon S, Bose S, Hur J, Jun K, Kim YK, Cho KS, Koo BS (2011) A modified formulation of Chinese traditional medicine improves memory impairment and reduces Aβ level in the Tg-APPswe/PS1dE9 mouse model of Alzheimer’s disease. J Ethnopharmacol 137:783-789.
Kautu BB, Carrasquilla A, Hicks ML, Caldwell KA, Caldwell GA (2013) Valproic acid ameliorates C. elegans dopaminergic neurodegeneration with implications for ERK-MAPK signaling. Neurosci Lett 541:116-119.
Keowkase R, Aboukhatwa M, Luo Y (2010) Fluoxetine protects against amyloid-beta toxicity, in part via daf-16 mediated cell signaling pathway, in Caenorhabditis elegans. Neuropharmacology 59:358-365.
Kessing LV, Søndergård L, Forman JL, Andersen PK (2008) Lithium treatment and risk of dementia. Arch Gen Psychiatry 65:1331-1335.
Khan A, Jamwal S, Bijjem KR, Prakash A, Kumar P (2015) Neuroprotective effect of hemeoxygenase-1/glycogen synthase kinase-3β modulators in 3-nitropropionic acid-induced neurotoxicity in rats. Neuroscience 287:66-77.
Kidd SK, Schneider JS (2011) Protective effects of valproic acid on the nigrostriatal dopamine system in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience 194:189-194.
Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, Rumbaugh G (2010) Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 35:870-880.
Kim YH, Rane A, Lussier S, Andersen JK (2011) Lithium protects against oxidative stress-mediated cell death in α-synuclein-overexpressing in vitro and in vivo models of Parkinson’s disease. J Neurosci Res 89:1666-1675.
Kohl Z, Winner B, Ubhi K, Rockenstein E, Mante M, Münch M, Barlow C, Carter T, Masliah E, Winkler J (2012) Fluoxetine rescues impaired hippocampal neurogenesis in a transgenic A53T synuclein mouse model. Eur J Neurosci 35:10-19.
Kontopoulos E, Parvin JD, Feany MB (2006) α-Synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet 15:3012-3023.
Lauterbach EC (2012a) Psychotropic drug effects on gene transcriptomics relevant to Alzheimer disease. Alzheimer Dis Assoc Disord 26:1-7.
Lauterbach EC (2012b) Psychotropic drug effects on gene transcriptomics relevant to Parkinson’s disease. Prog Neuropsychopharmacol Biol Psychiatry 38:107-115.
Lauterbach EC (2013a) Psychotropics regulate Skp1a, Aldh1a1, and Hspa8 transcription—potential to delay Parkinson’s disease. Prog Neuropsychopharmacol Biol Psychiatry 40:236-239.
Lauterbach EC (2013b) Neuroprotective effects of psychotropic drugs in Huntington’s disease. Int J Mol Sci 14:22558-22603.
Lauterbach EC (2016) Repurposing psychiatric medicines to target activated microglia in anxious mild cognitive impairment and early Parkinson’s disease. Am J Neurogener Dis 5:29-51.
Lauterbach EC, Mendez MF (2011) Psychopharmacological neuroprotection in neurodegenerative diseases, part III: criteria-based assessment: a report of the ANPA committee on research. J Neuropsychiatry Clin Neurosci 23:242-260.
Lauterbach EC, Victoroff J, Coburn KL, Shillcutt SD, Doonan SM, Mendez MF (2010) Psychopharmacological neuroprotection in neurodegenerative disease: assessing the preclinical data. J Neuropsychiatry Clin Neurosci 22:8-18.
Lee ST, Chu K, Park JE, Kang L, Ko SY, Jung KH, Kim M (2006) Memantine reduces striatal cell death with decreasing calpain level in 3-nitropropionic model of Huntington’s disease. Brain Res 1118:199-207.
Leonard DP, Kidson MA, Brown JG, Shannon PJ, Taryan S (1975) A double blind trial of lithium carbonate and haloperidol in Huntington’s chorea. Aust N Z J Psychiatry 9:115-118.
Li XZ, Chen XP, Zhao K, Bai LM, Zhang H, Zhou XP (2013) Therapeutic effects of valproate combined with lithium carbonate on MPTP-induced parkinsonism in mice: possible mediation through enhanced autophagy. Int J Neurosci 123:73-79.
Lieu CA, Dewey CM, Chinta SJ, Rane A, Rajagopalan S, Batir S, Kim YH, Andersen JK (2014) Lithium prevents parkinsonian behavioral and striatal phenotypes in an aged parkin mutant transgenic mouse model. Brain Res 1591:111-117.
Loewenstein DA, Curiel RE, Greig MT, Bauer RM, Rosado M, Bowers D, Wicklund M, Crocco E, Pontecorvo M, Joshi AD, Rodriguez R, Barker WW, Hidalgo J, Duara R (2016) A novel cognitive stress test for the detection of preclinical Alzheimer disease: discriminative properties and relation to amyloid load. Am J Geriatr Psychiatry 24:804-813.
Lu PH, Edland SD, Teng E, Tingus K, Petersen RC, Cummings JL; Alzheimer’s Disease Cooperative Study Group (2009) Donepezil delays progression to AD in MCI subjects with depressive symptoms. Neurology 72:2115-2121.
Luo G, Liu M, He J, Guo H, Xue M, Wang X, Li XM (2014) Quetiapine attenuates recognition memory impairment and hippocampal oxidative stress in a transgenic mouse model of Alzheimer’s disease. Neuroreport 25:647-650.
Mahlknecht P, Seppi K, Poewe W (2015) The concept of prodromal Parkinson’s disease. J Parkinson’s Dis 5:681-697.
Martinez-Coria H, Green KN, Billings LM, Kitazawa M, Albrecht M, Rammes G, Parsons CG, Gupta S, Banerjee P, LaFerla FM (2010) Memantine improves cognition and reduces Alzheimer’s-like neuropathology in transgenic mice. Am J Pathol 176:870-880.
McBride SM, Choi CH, Schoenfeld BP, Bell AJ, Liebelt DA, Ferreiro D, Choi RJ, Hinchey P, Kollaros M, Terlizzi AM, Ferrick NJ, Koenigsberg E, Rudominer RL, Sumida A, Chiorean S, Siwicki KK, Nguyen HT, Fortini ME, McDonald TV, Jongens TA (2010) Pharmacological and genetic reversal of age-dependent cognitive deficits attributable to decreased presenilin function. J Neurosci 30:9510-9522.
Monti B, Gatta V, Piretti F, Raffaelli SS, Virgili M, Contestabile A (2010) Valproic acid is neuroprotective in the rotenone rat model of Parkinson’s disease: involvement of alpha-synuclein. Neurotox Res 17:130-141.
Morris G, Berk M (2016) The putative use of lithium in Alzheimer’s disease. Curr Alzheimer Res 13:853-861.
Motoi Y, Shimada K, Ishiguro K, Hattori N (2014) Lithium and autophagy. ACS Chem Neurosci 5:434-442.
Mowla A, Mosavinasab M, Pani A (2007) Does fluoxetine have any effect on the cognition of patients with mild cognitive impairment? A double-blind, placebo-controlled, clinical trial. J Clin Psychopharmacol 27:67-70.
Nakamura S, Murayama N, Noshita T, Katsuragi R, Ohno T (2006) Cognitive dysfunction induced by sequential injection of amyloid-beta and ibotenate into the bilateral hippocampus; protection by memantine and MK-801. Eur J Pharmacol 548:115-122.
Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K (2005) Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A 102:6990-6995.
Noh H, Seo H (2014) Age-dependent effects of valproic acid in Alzheimer’s disease (AD) mice are associated with nerve growth factor (NGF) regulation. Neuroscience 266:255-265.
Nunes MA, Schöwe NM, Monteiro-Silva KC, Baraldi-Tornisielo T, Souza SI, Balthazar J, Albuquerque MS, Caetano AL, Viel TA, Buck HS (2015) Chronic microdose lithium treatment prevented memory loss and neurohistopathological changes in a transgenic mouse model of Alzheimer’s disease. PLoS One 10:e0142267.
Nunes MA, Viel TA, Buck HS (2013) Microdose lithium treatment stabilized cognitive impairment in patients with Alzheimer’s disease. Curr Alzheimer Res 10:104-107.
Nunes PV, Forlenza OV, Gattaz WF (2007) Lithium and risk for Alzheimer’s disease in elderly patients with bipolar disorder. Br J Psychiatry 190:359-360.
Nyakas C, Granic I, Halmy LG, Banerjee P, Luiten PG (2011) The basal forebrain cholinergic system in aging and dementia. Rescuing cholinergic neurons from neurotoxic amyloid-ß42 with memantine. Behav Brain Res 221:594-603.
Obermayr RP, Mayerhofer L, Knechtelsdorfer M, Mersich N, Huber ER, Geyer G, Tragl KH (2005) The age-related down-regulation of the growth hormone/insulin-like growth factor-1 axis in the elderly male is reversed considerably by donepezil, a drug for Alzheimer’s disease. Exp Gerontol 40:157-163.
Ondo WG, Mejia NI, Hunter CB (2007) A pilot study of the clinical efficacy and safety of memantine for Huntington’s disease. Parkinsonism Relat Disord 13:453-454.
Peters O, Lorenz D, Fesche A, Schmidtke K, Hüll M, Perneczky R, Rüther E, Möller HJ, Jessen F, Maier W, Kornhuber J, Jahn H, Luckhaus C, Gertz HJ, Schröder J, Pantel J, Teipel S, Wellek S, Frölich L, Heuser I (2012) A combination of galantamine and memantine modifies cognitive function in subjects with amnestic MCI. J Nutr Health Aging 16:544-548.
Petersen RC, Thomas RG, Grundman M, Bennett D, Doody R, Ferris S, Galasko D, Jin S, Kaye J, Levey A, Pfeiffer E, Sano M, van Dyck CH, Thal LJ; Alzheimer’s Disease Cooperative Study Group (2005) Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med 352:2379-2388.
Piersanti M, Capannolo M, Turchetti M, Serroni N, De Berardis D, Evangelista P, Costantini P, Orsini A, Rossi A, Maggio R (2014) Increase in mortality rate in patients with dementia treated with atypical antipsychotics: a cohort study in outpatients in Central Italy. Riv Psichiatr 49:34-40.
Pouladi MA, Brillaud E, Xie Y, Conforti P, Graham RK, Ehrnhoefer DE, Franciosi S, Zhang W, Poucheret P, Compte E, Maurel JC, Zuccato C, Cattaneo E, Néri C, Hayden MR (2012) NP03, a novel low-dose lithium formulation, is neuroprotective in the YAC128 mouse model of Huntington disease. Neurobiol Dis 48:282-289.
Qing H, He G, Ly PT, Fox CJ, Staufenbiel M, Cai F, Zhang Z, Wei S, Sun X, Chen CH, Zhou W, Wang K, Song W (2008) Valproic acid inhibits Abeta production, neuritic plaque formation, and behavioral deficits in Alzheimer’s disease mouse models. J Exp Med 205:2781-2789.
Reilmann R, Leavitt BR, Ross CA (2014) Diagnostic criteria for Huntington’s disease based on natural history. Mov Disord 29:1335-1341.
Salloway S, Correia S, Richardson S (2008) Key lessons learned from short-term treatment trials of cholinesterase inhibitors for amnestic MCI. Int Psychogeriatr 20:40-46.
Salloway S, Ferris S, Kluger A, Goldman R, Griesing T, Kumar D, Richardson S; Donepezil 401 Study Group (2004) Efficacy of donepezil in mild cognitive impairment: a randomized placebo-controlled trial. Neurology 63:651-657.
Sarkar S, Krishna G, Imarisio S, Saiki S, O’Kane CJ, Rubinsztein DC (2008) A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum Mol Genet 17:170-178.
Schneider LS, Dagerman KS, Insel P (2005) Risk of death with atypical antipsychotic drug treatment for dementia: meta-analysis of randomized placebo-controlled trials. JAMA 294:1934-1943.
Scholtzova H, Wadghiri YZ, Douadi M, Sigurdsson EM, Li YS, Quartermain D, Banerjee P, Wisniewski T (2008) Memantine leads to behavioral improvement and amyloid reduction in Alzheimer’s-disease-model transgenic mice shown as by micromagnetic resonance imaging. J Neurosci Res 86:2784-2791.
Schuff N, Suhy J, Goldman R, Xu Y, Sun Y, Truran-Sacrey D, Murthy A (2011) An MRI substudy of a donepezil clinical trial in mild cognitive impairment. Neurobiol Aging 32:2318.e31-41.
Shi J, Tian J, Zhang X, Zeng C, Wei M, Wang P, Wang Y (2013) A combination extract of Renshen (Panax Ginseng), Yinyanghuo (Herba Epimedii Brevicornus), Yuanzhi (Radix Palygalae) and Jianghuang (Rhizoma Curcumae Longae) decreases glycogen synthase kinase 3beta expression in brain cortex of APPV7171 transgenic mice. J Tradit Chin Med 33:211-217.
Sofola O, Kerr F, Rogers I, Killick R, Augustin H, Gandy C, Allen MJ, Hardy J, Lovestone S, Partridge L (2010) Inhibition of GSK-3 ameliorates Abeta pathology in an adult-onset Drosophila model of Alzheimer’s disease. PLoS Genet 6:e1001087.
Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR Jr., Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, et al. (2011) Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dementia 7:280-292.
Stip E, Dufresne J, Lussier I, Yatham L (2000) A double-blind, placebo-controlled study of the effects of lithium on cognition in healthy subjects: mild and selective effects on learning. J Affect Disord 60:147-157.
Tariot PN, Schneider LS, Cummings J, Thomas RG, Raman R, Jakimovich LJ, Loy R, Bartocci B, Fleisher A, Ismail MS, Porsteinsson A, Weiner M, Jack CR Jr, Thal L, Aisen PS; Alzheimer’s Disease Cooperative Study Group (2011) Chronic divalproex sodium to attenuate agitation and clinical progression of Alzheimer disease. Arch Gen Psychiatry 68:853-861.
Tariot PN, Aisen PS (2009) Can lithium or valproate untie tangles in Alzheimer’s disease? J Clin Psychiatry 70:919-921.
Tempier A, He J, Zhu S, Zhang R, Kong L, Tan Q, Luo H, Kong J, Li XM (2013) Quetiapine modulates conditioned anxiety and alternation behavior in Alzheimer’s transgenic mice. Curr Alzheimer Res 10:199-206.
Terao T, Nakano H, Inoue Y, Okamoto T, Nakamura J, Iwata N (2006) Lithium and dementia: a preliminary study. Prog Neuropsychopharmacol Biol Psychiatry 30:1125-1128.
Trujillo-Estrada L, Jimenez S, De Castro V, Torres M, Baglietto-Vargas D, Moreno-Gonzalez I, Navarro V, Sanchez-Varo R, Sanchez-Mejias E, Davila JC, Vizuete M, Gutierrez A, Vitorica J (2013) In vivo modification of Abeta plaque toxicity as a novel neuroprotective lithium-mediated therapy for Alzheimer’s disease pathology. Acta Neuropathol Commun 1:73.
Van Dam D, Coen K, De Deyn PP (2008) Cognitive evaluation of disease-modifying efficacy of donepezil in the APP23 mouse model for Alzheimer’s disease. Psychopharmacology (Berl) 197:37-43.
Van Dam D, De Deyn PP (2006) Cognitive evaluation of disease-modifying efficacy of galantamine and memantine in the APP23 model. Eur Neuropsychopharmacol 16:59-69.
Vestergaard P, Baastrup PC, Petersson H (1977) Lithium treatment of Huntington’s chorea. A placebo-controlled clinical trial. Acta Psychiatr Scand 56:183-188.
Voisine C, Varma H, Walker N, Bates EA, Stockwell BR, Hart AC (2007) Identification of potential therapeutic drugs for Huntington’s disease using Caenorhabditis elegans. PLoS One 2:e504.
Wang J, Zhang Y, Xu H, Zhu S, Wang H, He J, Zhang H, Guo H, Kong J, Huang Q, Li XM (2014) Fluoxetine improves behavioral performance by suppressing the production of soluble β-amyloid in APP/ PS1 mice. Curr Alzheimer Res 11:672-680.
Wei H, Qin ZH, Senatorov VV, Wei W, Wang Y, Qian Y, Chuang DM (2001) Lithium suppresses excitotoxicity-induced striatal lesions in a rat model of Huntington’s disease. Neuroscience 106:603-612.
Weintraub D, Chiang C, Kim HM, Wilkinson J, Marras C, Stanislawski B, Mamikonyan E, Kales HC (2016) Association of antipsychotic use with mortality risk in patients with Parkinson disease. JAMA Neurol 73:535-541.
Wood NI, Morton AJ (2003) Chronic lithium chloride treatment has variable effects on motor behaviour and survival of mice transgenic for the Huntington’s disease mutation. Brain Res Bull 61:375-383.
Wu CR, Lin HC, Su MH (2014) Reversal by aqueous extracts of Cistanche tubulosa from behavioral deficits in Alzheimer’s disease-like rat model: relevance for amyloid deposition and central neurotransmitter function. BMC Complement Altern Med 14:202.
Wu YY, Wang X, Tan L, Liu D, Liu XH, Wang Q, Wang JZ, Zhu LQ (2013) Lithium attenuates scopolamine-induced memory deficits with inhibition of GSK-3β and preservation of postsynaptic components. J Alzheimers Dis 37:515-527.
Ximenes JC, Neves KR, Leal LK, do Carmo MR, Brito GA, Naffah-Mazzacoratti Mda G, Cavalheiro ÉA, Viana GS (2015) Valproic acid neuroprotection in the 6-OHDA model of Parkinson’s disease is possibly related to its anti-inflammatory and HDAC inhibitory properties. J Neurodegener Dis 2015:313702.
Yong Y, Ding H, Fan Z, Luo J, Ke ZJ (2011) Lithium fails to protect dopaminergic neurons in the 6-OHDA model of Parkinson’s disease. Neurochem Res 36:367-374.
Youdim MB, Arraf Z (2004) Prevention of MPTP (N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) dopaminergic neurotoxicity in mice by chronic lithium: involvements of Bcl-2 and Bax. Neuropharmacology 46:1130-1140.
Zádori D, Geisz A, Vámos E, Vécsei L, Klivényi P (2009) Valproate ameliorates the survival and the motor performance in a transgenic mouse model of Huntington’s disease. Pharmacol Biochem Behav 94:148-153.
Zhang X, Heng X, Li T, Li L, Yang D, Zhang X, Du Y, Doody RS, Le W (2011) Long-term treatment with lithium alleviates memory deficits and reduces amyloid-ßß production in an aged Alzheimer’s disease transgenic mouse model. J Alzheimers Dis 24:739-749.
Zhu S, Shi R, Li V, Wang J, Zhang R, Tempier A, He J, Kong J, Wang JF, Li XM (2014) Quetiapine attenuates glial activation and proinflammatory cytokines in APP/PS1 transgenic mice via inhibition of nuclear factor-κB pathway. Int J Neuropsychopharmacol 18:pii:pyu022.
Zhu S, He J, Zhang R, Kong L, Tempier A, Kong J, Li XM (2013) Therapeutic effects of quetiapine on memory deficit and brain β-amyloid plaque pathology in a transgenic mouse model of Alzheimer’s disease. Curr Alzheimer Res 10:270-278.
Zimmer ER, Kalinine E, Haas CB, Torrez VR, Souza DO, Muller AP, Portela LV (2012) Pretreatment with memantine prevents Alzheimer-like alterations induced by intrahippocampal okadaic acid administration in rats. Curr Alzheimer Res 9:1182-1190.
*Correspondence to: Edward C. Lauterbach, M.D., F.A.N.P.A., D.F.A.P.A.,
eclbgnp@earthlink.net.
10.4103/1673-5374.194708
Accepted: 2016-10-17
猜你喜欢
杂志排行

中国神经再生研究(英文版)的其它文章
- The status of Nrf2-based therapeutics: current perspectives and future prospects
- The Epigenome as a therapeutic target for Parkinson’s disease
- Neuroinflammation, neurodegeneration and regeneration in multiple sclerosis: intercorrelated manifestations of the immune response
- Applicability of tooth derived stem cells in neural regeneration
- Cortical spreading depression-induced preconditioning in the brain
- Targeting neuronal nitric oxide synthase as a valuable strategy for the therapy of neurological disorders