10个菲律宾蛤仔野生群体的遗传多样性研究*
2016-01-15胡利莎马培振王海艳
胡利莎 张 振 马培振 王海艳①
(1. 中国科学院海洋研究所 青岛 266071; 2. 中国科学院大学 北京 100049; 3. 中国海洋大学 青岛 266003)
菲律宾蛤仔(Ruditapes philippinarum)属于软体动物门(Mollusca), 双壳纲(Bivalvia), 帘蛤目(Venerioda), 帘蛤科(Veneridae), 蛤仔属(Ruditapes)。菲律宾蛤仔分布于俄罗斯、日本、菲律宾和中国沿海,在我国河北、山东、浙江、福建、广东等省区沿海分布较广。菲律宾蛤仔生长迅速, 适应性强, 肉味鲜,是我国4大养殖贝类之一, 具有较高的经济价值。近些年来, 我国菲律宾蛤仔的人工养殖规模快速发展,异地引种养殖现象频繁出现, 这一人类活动使菲律宾蛤仔野生群体间的遗传多样性结构受到了一定的影响, 因此, 研究我国菲律宾蛤仔的种群遗传学, 不仅能够保护菲律宾蛤仔的种质资源, 还能为菲律宾蛤仔养殖业的发展提供良好的生物学依据。
目前对菲律宾蛤仔种群遗传学已有较多研究。裴赢等(2006)采用RAPD分子标记技术分析了大连沿海6个菲律宾蛤仔种群的遗传多样性及其分布, 结果表明该地区的菲律宾蛤仔种群具有较高的遗传多样性,且种群之间存在一定数量的遗传变异。葛京盈等(2008)研究表明大连、丹东、青岛和锦州4群体生化遗传特征上未见明显差异, 且4个野生群体的遗传多样性属偏低水平。Bi等(2012)研究结果显示烟台和威海的菲律宾蛤仔无明显的种群结构, 且具有较高的单倍型多样性。Ren等(2006)利用同工酶的方法、李旭光等(2009)采用聚丙稀酰胺同工酶技术对菲律宾蛤仔不同群体遗传特征进行了研究, 认为中国沿海菲律宾蛤仔分化成南、北两大类群。Sekine等(2006)和Mao等(2011)利用COI基因的研究也得出类似的结果。
结合前人的研究结果可以看出, 关于北方菲律宾蛤仔的遗传多样性的研究结果各不相同。Ren等(2006)未对中国沿海南北两大类群的遗传多样性进行详细的分析。在此基础上本研究将结合 COI和 16S rRNA两种基因分析中国沿海菲律宾蛤仔的遗传多样性和系统进化关系, 为菲律宾蛤仔种群的遗传结构特征以及生物资源保护提供更加准确的分子生物学信息。
1 材料与方法
1.1 材料与样品制备
实验所需样品分别从大连、荣成、连云港、霞浦、莆田、广州、阳江、湛江、钦州、儋州当地采集, 每群体个体数量为 12—23个, 样品采集后立即用 95%酒精固定, 保存备用。图1为样品采集地的分布。

图1 菲律宾蛤仔样品分布图Fig.1 Map of sampling sites for R. philippinarum
1.2 DNA的提取、扩增及测序
取菲律宾蛤仔闭壳肌肌肉约 100mg充分剪碎,用TIANamp海洋动物DNA提取试剂盒(北京天根生物有限公司)提取全基因组。
线粒体COI基因扩增引物LCO1490 (5′-GGTCA ACAAATCATAAAGATATTGG-3′)和 HCO2198 (5′-A AACTTCAGGGTGACCAAAAAATCA-3′) (Folmeret al, 1994); 16S rRNA基因扩增引物16S rRNAar (5′-CGCCTGTTTATCAAAAACAT-3′) 和 16S rRNAbr(5′-CCGGTCTGAACTCAGMTCAYG-3′) (Anderson,2000), 由上海桑尼生物科技有限公司合成。实验中PCR 反应体系: 2μL 10×PCR buffer, 1.5mM Mg2+,200μM dNTPs, 1U Taq DNA polymerase, 正、反向引物各 0.4μM, 2μL 模板 DNA 溶液(10–100ng/μL), 用超纯水补充至 25μL。PCR扩增反应程序为: 预变性94°C 5min; 94°C 30s, 48–52°C 1min, 72°C 1min, 循环30–35 次; 总延伸 72°C 10min。
PCR产物经1.5%–2.0%琼脂糖电泳凝胶纯化后送上海桑尼生物科技有限公司测序, COI基因测序引物用LCO1490, 16S rRNA基因测序引物用16S rRNar。
1.3 数据分析
单倍型: Haplotype, 指若干个决定同一性状的紧密连锁的基因构成的基因型, 及一条染色体上 SNP基因型的组合。
DNA序列结果由CLUSTAL X软件(Thompsonet al, 1997)采用默认参数对位, 并进一步经人工校对。对位后将碱基序列输入 DnaSP 5软件(Rozasetal,2003)统计单倍型, 并统计单倍型在各采样点的分布情况, 并对多态位点数、单倍型数、核苷酸多样性指数(Pi)、单倍型多样性指数(Hd)、平均核苷酸差异数(K)等遗传多样性参数进行计算; 采用 Mega5.1(Tamuraetal, 2004)软件进行遗传距离计算和聚类分析, 并构建NJ进化树。采用IBDWS(Jensenetal, 2005)软件中的 Mantel检测评估菲律宾蛤仔群体的遗传距离和地理距离的相关性。运用 ARLEQUIN v3.5(Excoffieretal, 2005)软件中的分子变异分析(AMOVA)方法估算遗传变异在群体内和群体间的分布, 并计算群体间遗传分化系数(F-statistics,Fst)及其显著性(重复次数1000)。
2 结果
2.1 菲律宾蛤仔群体基因序列碱基组成
COI和16S rRNA基因序列通过BLAST 分析、比较确认, 经CLUSTAL X同源排序, 除去引物和部分端序列, 分别得到长度为632bp和473bp的基因片段。利用Mega5.1软件计算菲律宾蛤仔群体COI和16S rRNA基因序列碱基组成, 不同群体的各碱基组成基本一致,COI基因T、C、A、G、A+T和G+C的平均含量分别为41.4%, 12.8%, 23.4%, 22.3%, 64.8%, 35.2%, AT含量明显高于GC含量。16S rRNA基因T、C、A、G、A+T和G+C的平均含量分别为33.0%, 10.6%, 33.6%, 22.8%,66.6%, 33.4%, AT含量亦明显高于GC含量。
2.2 菲律宾蛤仔群体遗传多样性分析
利用DnaSP 5软件计算群体遗传多样性参数, 结果见表1和表2。根据COI基因序列计算遗传多样性参数, 在183个菲律宾蛤仔个体中, 共检测到67个单倍型, 且每个群体都具有各自的单倍型。群体特有单倍型50个, 占单倍型总数的74.63%; 共享单倍型17个, 占单倍型总数的25.37%, 其中共享单倍型Hap_4的个体数最多 31个, 占总个体数的 16.94%, 共享单倍型Hap_4在除大连群体和荣成群体外的8个群体中均有分布, 而Hap_16分布在大连群体和荣成群体中。COI基因共检测到 126个核苷酸多态位点, 包括 83个单突变位点, 43个简约信息位点, 总群体的平均核苷酸差异数为 6.35447, 核苷酸多样性指数为0.01005。其中荣成群体遗传多样性参数较高, 莆田群体遗传多样性参数较低。

表1 基于COI基因序列片段的10个菲律宾蛤仔群体的遗传多样性参数Tab.1 Genetic diversity parameters of partial COI gene in 10 populations of R. philippinarum

表2 基于16S rRNA基因序列片段的10个菲律宾蛤仔群体的遗传多样性参数Tab.2 Genetic diversity parameters of partial 16S rRNA gene in 10 populations of R. philippinarum
根据 16S rRNA 基因序列计算遗传多样性参数,在193个菲律宾蛤仔个体中, 共检测到了22个单倍型, 群体特有单倍型15个, 占单倍型总数的68.18%,共享单倍型7个, 占单倍型总数的31.82%, 其中共享单倍型Hap_1个体数最多138, 占总个体数的71.50%,在10个群体中均有分布。共检测到21个核苷酸多态位点, 包括14个单突变位点, 7个简约信息位点, 总群体的平均核苷酸差异数为 0.86161, 核苷酸多样性指数为 0.00182。其中荣成和大连群体遗传多样性参数普遍高于其余群体, 而霞浦群体因只有一种单倍型其遗传多样性参数为零。
2.3 菲律宾蛤仔群体的遗传距离和聚类分析
利用Mega5.1所计算的群体内、群体间遗传距离如表3和4所示。基于COI基因的计算结果: 荣成群体内遗传距离最高为0.014052, 而莆田群体内遗传距离最低为 0.005089, 说明荣成群体遗传多样性最高,莆田群体遗传多样性较低; 群体间遗传距离在 0.005—0.021, 其中荣成群体和大连群体与其他群体的遗传距离均较高; 荣成与其他群体间遗传距离中与钦州群体的最高, 与大连群体的最低; 钦州和广州、儋州、连云港、霞浦、莆田、湛江、阳江8个群体两两间的遗传距离均低于与荣成和大连之间的遗传距离。基于16S rRNA基因计算的结果: 荣成群体内遗传距离最高为0.002938, 而霞浦群体内遗传距离最低为0,说明荣成群体遗传多样性最高, 霞浦群体遗传多样性较低; 群体间遗传距离在 0.000151—0.004378, 其中荣成群体和大连群体与其他群体的遗传距离均较高; 荣成与其他群体间遗传距离中与连云港群体的最高, 与大连群体的最低; 钦州和广州、儋州、连云港、霞浦、莆田、湛江、阳江8个群体两两间的遗传距离均低于与荣成和大连之间的遗传距离。
利用 Mega5.1软件中的 NJ法对菲律宾蛤仔 10个群体进行系统树的构建, 群体之间的系统聚类图如图2和3所示, 从图中可以看出, 基于COI基因和16S rRNA基因构建的NJ树均分为两大支, 大连、荣成群体聚为一支, 其余8个群体聚为一支。
对菲律宾蛤仔10个群体进行地理距离和遗传距离的 Mantel测试, 发现菲律宾蛤仔群体的遗传距离与地理距离之间不存在明显的线性关系(COI基因:R=0.5430,P=0.9874; 16S rRNA 基因:R=0.6395,P=0.9956)(图 4、图 5)。
2.4 菲律宾蛤仔群体的遗传分化
运用ARLEQUIN v3.5计算的群体间遗传分化系数(Fst)及其显著性如表3和表4所示。由表3可看出,基于COI基因所得出的10个群体间的遗传分化指数在–0.03146—0.49684之间, 其中湛江与莆田之间的遗传分化系数最小, 大连与莆田之间的遗传分化系数最大。群体间两两比对的组中, 18个组间的遗传分化表现为差异不显著(P>0.05), 10个组表现为显著差异(P<0.05), 17个组表现为极显著差异(P<0.01), 除大连和荣成群体外的其他群体相互之间均未达到分化极显著(除霞浦与广州群体间的遗传分化极显著外)。由表4可看出, 基于16S rRNA基因所得出的10个群体间的遗传分化指数在–0.04027—0.63743之间, 其中荣成与霞浦之间的遗传分化指数最大, 荣成与大连之间的遗传分化指数最小。群体间两两比对的组中,22个组间的Fst值统计检验结果为差异不显著(P>0.05), 4个组为差异显著(P<0.05), 19个组表现为差异极显著(P<0.01), 连云港群体与钦州、儋州、霞浦、湛江群体间的遗传分化显著, 与其余群体极显著;结合COI基因和16S rRNA基因总体来看, 荣成群体除与大连群体间的遗传分化差异不显著外, 与其他群体间的遗传分化均为极显著, 并且大连群体也是除与荣成群体间的遗传分化不显著外, 与其他群体间的遗传分化均为极显著; 群体间遗传分化显著或者是极显著的均出现在荣成群体、大连群体、连云港群体与其他群体之间。按照 NJ树的分组, 将荣成群体和大连群体分为一组, 其余8个群体分为一组进行Fst分析, 结果显示基于COI基因和16S rRNA基因组群间的遗传分化指数分别为 0.05615, 差异极显著(P<0.01)和 0.37253, 差异极显著(P<0.01)。

表3 基于COI基因序列片段的菲律宾蛤仔群体间遗传距离(对角线下), 群体内遗传距离(粗线显示)和群体间分化系数(对角线上)Tab.3 Genetic distances between populations (below the diagonal), genetic distances within populations (in bold values) and pairwise Fst (above the diagonal) of 10 populations of R. philippinarum based on partial COI gene

表4 基于16S rRNA基因序列片段的菲律宾蛤仔群体间遗传距离(对角线下), 群体内遗传距离(粗线显示)和群体间分化系数(对角线上)Tab.4 Genetic distances between populations (below the diagonal), genetic distances within populations (in bold values) and pairwise Fst (above the diagonal) of 10 populations of R. philippinarum based on partial 16S rRNA gene

图2 基于COI基因菲律宾蛤仔10个群体的NJ树Fig.2 NJ tree based on the COI gene of 10 populations of R.philippinarum
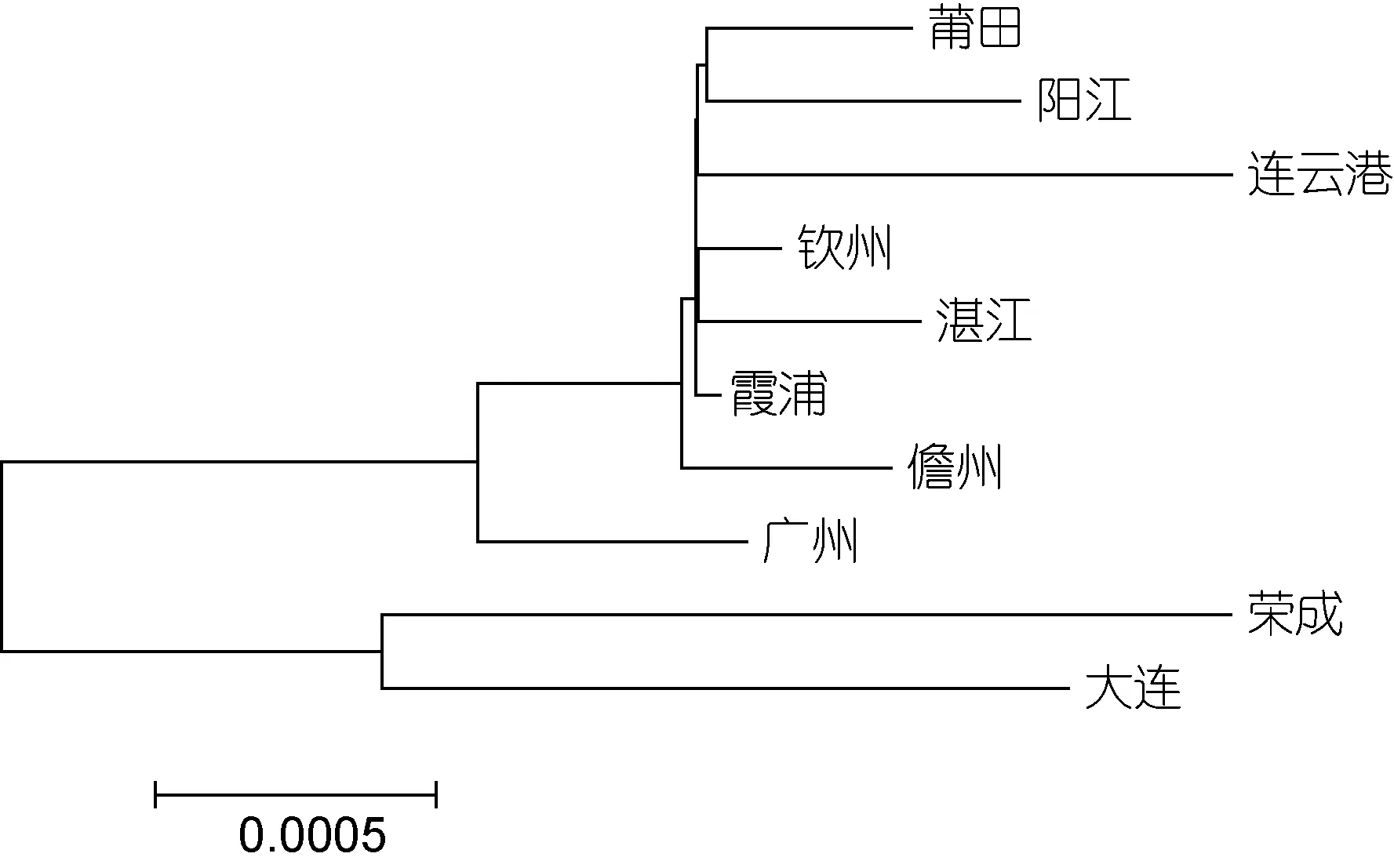
图3 基于16S rRNA基因菲律宾蛤仔10个群体的NJ树Fig.3 NJ tree based on the 16S rRNA of 10 populations of R.philippinarum
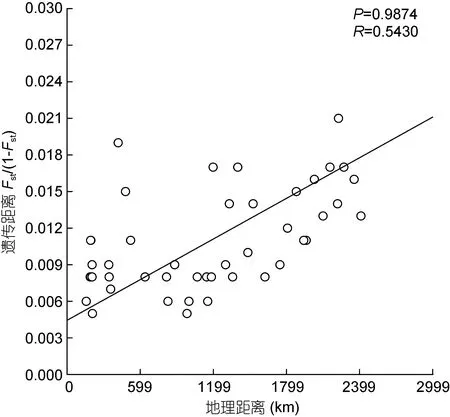
图4 菲律宾蛤仔COI基因的遗传距离模式(IBD)分析Fig.4 IBD analysis based on COI gene of R. philippinarum

图5 菲律宾蛤仔16S rRNA基因的遗传距离模式(IBD)分析Fig.5 IBD analysis based on the 16S rRNA of R. philippinarum
对菲律宾蛤仔10个群体进行AMOVA分析(如表5和6)表明, 基于COI基因群体内遗传变异为78.17%,群体间的遗传变异为21.83%, 基于16S rRNA基因群体内遗传变异为 66.86%, 群体间的遗传变异为33.14%, 说明菲律宾蛤仔群体遗传分化主要表现为群体内分化, 群体内部遗传多态性较高, 而在Fst分析中, 一般认为 0<Fst<0.05表示群体间无显著分化,0.05<Fst<0.15 表示群体间的分化程度中等,0.15<Fst<0.25 表示群体间的分化程度较大,Fst>0.25表示群体间的分化程度极大, 基于COI基因和16S rRNA基因菲律宾蛤仔群体总的遗传分化指数分别为0.21828和0.33143, 说明菲律宾蛤仔群体间存在较大的遗传分化。

表5 基于COI基因序列片段的菲律宾蛤仔群体遗传变异的AMOVA分析Tab.5 Results of hierarchical AMOVA based on the partial COI sequences of 10 populations of R. philippinarum

表6 基于16S rRNA基因序列片段的菲律宾蛤仔群体遗传变异的AMOVA分析Tab.6 Results of hierarchical AMOVA analysis based on the partial 16S rRNA sequences of the ten populations of R.philippinarum
3 讨论
线粒体基因是群体遗传学和进化生物学研究中的重要分子标记。线粒体基因上的不同片段具有不同的解析能力, 即使是同一基因在不同的物种间的解析能力也是不同的。一般而言, 16S rRNA和COI基因在同一种群中变异程度较低, 但在不同的种群之间16S rRNA基因较COI基因具有明显的保守性。本研究中菲律宾蛤仔10个群体16S rRNA和COI基因片段检测到的多态位点数分别为126和21, 核苷酸多样性指数(Pi)分别为0.01005和0.00182, 此结果说明菲律宾蛤仔的不同的群体之间16S rRNA基因较COI基因具有明显的保守性。
3.1 菲律宾蛤仔16S rRNA和COI基因的碱基组成和遗传多样性
本研究得到的10个菲律宾蛤仔群体的COI和16S rRNA两个基因片段各碱基组成基本一致, A+T平均含量分别为64.8%、66.6%, G+C平均含量35.2%、33.4%, 所有群体的A+T含量均显著高于G+C含量,这与其他贝类如文蛤属(文蛤、丽文蛤等)、泥蚶、太平洋牡蛎等线粒体基因的碱基组成相一致(Kongetal,2001; 潘宝平等, 2006; 郑文娟等, 2009)。
菲律宾蛤仔10个群体16S rRNA和COI基因片段单倍型多样性(Hd)分别为0.47674和0.941, 核苷酸多样性指数(Pi)分别为0.00182和0.01005, 与相近物种相比: 例如栉孔扇贝(Chlamys farreri)(刘亚军等,2002)、泥蚶(Tegillarca granosa)(郑文娟等, 2009)、光滑河篮(Potamocorbula laevis) (孙超等, 2013)、黑龙江河篮蛤(Potamocorbula amurensis)(孙超, 2013)、栉江珧(Atrina pectinta)(严加坤等, 2013)、厚壳贻贝(Mytilis coruscus)(杨振雄等, 2014), 表明我国沿海菲律宾蛤仔遗传多样性相对比较丰富, 大连群体遗传多样性较高, 裴赢等(2006)的研究结果也表明大连群体具有较高的遗传多样性。
3.2 菲律宾蛤仔的遗传距离和遗传分化
菲律宾蛤仔遗传参数Fst的 AMOVA分析表明:菲律宾蛤仔的遗传分化主要来自群体内(COI基因群体内变异贡献率为78.17%, 16S rRNA群体内变异贡献率为 66.86%), 说明群体内的遗传多态性较高; 另外, 依据各个群体间的 Fst分析, 荣成和大连群体间分化不显著, 而荣成、大连群体与其余8个群体间分化达到极显著水平(P<0.01)。根据遗传距离和遗传树的构建将菲律宾蛤仔 10个群体分为两组: 荣成群体和大连群体为一组, 其余8群体为一组, 基于两组进行的分析表明: 两组群间的遗传分化极显著, 因此可将我国沿海的菲律宾蛤仔分为南北两个类群, Ren等(2006)、李旭光等(2009)的研究结果也将我国沿海菲律宾蛤仔群体分为南北两个类群。
群体之间的遗传距离能反映群体间的亲缘关系。本研究结果表明荣成群体和大连群体与其他群体的遗传距离较远, 而其他群体间遗传距离较近, 所以荣成群体和大连群体亲缘关系较近, 其他8群体亲缘关系较近。亲缘关系的远近一般与地理位置远近一致,本研究中遗传距离与地理距离没有明显的相关性。如连云港群体没有与地理位置较近的荣成群体聚在一起, 反而与地理位置较远的南方群体聚在一起, 可能是越来越多的南方菲律宾蛤仔苗种被引入北方养殖,使得连云港本地的野生群体的遗传组成受到了影响,从而与南方种群结构越来越接近, 亲缘关系也越来越近; 也可能受沿岸流和暖流的影响, 使连云港以北和以南的基因交流受到了限制, 从而使南北群体产生了遗传分化。
Liu等(2007)研究结果表明异地购苗增养殖对菲律宾蛤仔野生群体的遗传结构产生了一定的影响,所以青岛群体与厦门群体的遗传相似度较高。但是本研究中大连群体和荣成群体具有较高的遗传相似度,而其余8群体具有较高的遗传相似度, 明显形成两大类群。虽然北方在养殖过程中很大一部分苗种引自南方, 但大连群体和荣成群体与南方种群未显示出高的遗传相似度, 说明大连和荣成的野生群体结构未被影响, 而其余8群体的遗传结构在一定程度上受到了苗种异地放养的影响。Kim等(2015)应用5个PNA探针很好地区别出了韩国当地菲律宾蛤仔和引自大连的菲律宾蛤仔。因此在本研究基础上, 如果能够应用 Kim 等(2015)的方法进一步对遗传结构受到苗种异地放养影响的群体进行研究, 确定这些群体受苗种异地放养影响的程度, 将为菲律宾蛤仔的种群结构研究提供更为有力的数据支撑。
4 结论
我国沿海菲律宾蛤仔种群具有相对较高的遗传多样性, 并且以连云港为界分为南北两大类群, 其中北方种群生物多样性高于南方种群, 本研究为菲律宾蛤仔的养殖和生物资源保护提供了生物学参考依据。至2014年, 10m等深线以内的浅海基本都被开发用于贝类的养殖, 而菲律宾蛤仔已成为浅海养殖第一大品种(车向庆等, 2015), 作为具有经济价值的物种, 为保证其生物多样性, 防止种质退化, 在养殖过程中要针对南北两大类群特点进行合理有效的保护。
车向庆, 冷忠业, 吴庆东等, 2015. 菲律宾蛤仔浅海养殖技术.科学养鱼, (11): 44
刘亚军, 喻子牛, 姜艳艳等, 2002. 栉孔扇贝16S rRNA基因片段序列的多态性研究. 海洋与湖沼, 33(5): 477—483
孙 超, 2013. 河蓝蛤属贝类生物学性状及基于线粒体 DNA的分子遗传学研究. 上海: 上海海洋大学硕士学位论文,23—28
孙 超, 刘志鸿, 杨爱国等, 2013. 光滑河蓝蛤 3个野生群体线粒体 COI基因遗传多样性研究. 湖南农业科学, (7):4—7
严加坤, 杨爱国, 周丽青等, 2013. 基于线粒体16S rRNA基因研究5个栉江珧野生群体的遗传多样性. 海洋科学, 37(2):36—42
李旭光, 许广平, 阎斌伦等, 2009. 菲律宾蛤仔不同地理群体生化遗传结构与变异的研究. 海洋科学, 33(4): 61—65
杨振雄, 毛阳丽, 宋 娜等, 2014. 浙江和福建沿海厚壳贻贝Mytilus coruscus群体的 COI序列比较分析. 海洋湖沼通报, (2): 82—88
郑文娟, 朱世华, 沈锡权等, 2009. 基于线粒体 COI基因序列探讨泥蚶的遗传分化. 动物学研究, 30(1): 17—23
葛京盈, 刘 萍, 高天翔, 2008. 菲律宾蛤仔 4个野生群体的同工酶分析. 海洋水产研究, 29(6): 63—70
裴 赢, 王晓红, 张恒庆, 2006. 大连渤海沿海菲律宾蛤仔种群的RAPD分析. 水产科学, 25(5): 250—252
潘宝平, 吴 琪, 张素萍等, 2006. 文蛤属 (Meretrix) 16S rRNA基因及ITS1序列的系统学分析. 海洋与湖沼, 37(4):342—347
Anderson F E, 2000. Phylogeny and historical biogeography of the loliginid squids (Mollusca: Cephalopoda) based on mitochondrial DNA sequence data. Molecular Phylogenetics and Evolution, 15(2): 191—214
Bi X X, Huang L, Jing M D et al, 2012. Analysis on genetic diversity of mitochondria from Venerupis philippinarum in the coasts of Yantai and Weihai. Agricultural Science &Technology-Hunan, 13(1): 32—35
Excoffier L, Laval G, Schneider S, 2005. Arlequin version 3.01:An integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online, 1: 47—50
Folmer O, Black M, Hoeh W et al, 1994. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology, 3(5): 294—299
Jensen J L, Bohonak A J, Kelley S T, 2005. Isolation by distance,web service. BMC Genetics, 6: 13
Kim E-M, Song M S, Hur D H et al, 2015. Easy method for discriminating the origins of manila clam Ruditapes philippinarum with a dual-labelled PNA-probe-based melting curve analysis. BioChip Journal, 9(3): 247—258
Kong X Y, Zhang L S, Yu Z N et al, 2002. Sequencing of ribosomal internal transcribed spacer regions and mitochondrial gene fragments in Crassostrea gigas. Journal of Fishery Sciences of China, 9(4): 304—308
Liu X Q, Bao Z M, Hu J J, et al, 2007. AFLP analysis revealed differences in genetic diversity of four natural populations of Manila clam (Ruditapes philippinarum) in China. Acta Oceanologica Sinica, 26(1): 150—158
Mao Y L, Gao T X, Yanagimoto T et al, 2011. Molecular phylogeography of Ruditapes philippinarum in the Northwestern Pacific Ocean based on COI gene. Journal of Experimental Marine Biology and Ecology, 407(2): 171—181 Ren Y P, Gao T X, Yang T Y, 2006. Isozyme analysis on the populations of Ruditapes philippinarum. Journal of Ocean University of China, 5(1): 58—62
Rozas J, Sánchez-DelBarrio J C, Messeguer X et al, 2003. DnaSP,DNA polymorphism analyses by the coalescent and other methods. Bioinformatics, 19(18): 2496—2497
Sekine Y, Yamakawa H, Takazawa S et al, 2006. Geographic variation of the COX1 gene of the short-neck clam Ruditapes philippinarum in coastal regions of Japan and China. Venus, 65: 229—240
Tamura K, Nei M, Kumar S, 2004. Prospects for inferring very large phylogenies by using the neighbor-joining method.Proceedings of the National Academy of Sciences of the United States of America, 101(30): 11030—11035
Thompson J D, Gibson T J, Plewniak F et al, 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools.Nucleic Acids Research, 25(24): 4876—4882
