有氧运动改善心肌梗死心脏交感神经重构的抗炎机制
2016-01-12邵承颖苏冉
邵承颖苏冉
摘要:目的:探讨有氧运动是否通过抑制NF-κB通路激活改善心肌梗死后交感神经重构。方法:45只健康雄性SD大鼠随机分为假手术组、心肌梗死组、心肌梗死+有氧运动组。结扎冠状动脉左前降支制备大鼠心肌梗死的模型,术后1周心肌梗死+有氧运动组成活大鼠进行4周跑台运动。采用免疫组织化学方法观察心肌梗死灶周及左室游离壁TH阳性神经纤维分布及表达;采用western blot检测心肌中NF-κB p65及IκBα蛋白表达;采用实时定量RT-PCR检测IL-1β及TNF-α mRNA表达。结果:与假手术组大鼠相比,心肌梗死组心肌中TH阳性神经纤维密度明显增加,形态粗大且空间分布紊乱,尤以梗死灶周为著;有氧运动干预后TH阳性神经纤维密度显著降低,形态更趋于正常。心肌梗死可激活NF-κB通路,表现为心肌核蛋白中NF-κB p65含量增加,胞质蛋白中IκBα含量降低,其下游转录产物IL-1β及TNF-α mRNA明显上调;而有氧运动干预可明显抑制NF-κB通路激活,减少NF-κB p65及增加IκBα蛋白表达,并下调IL-1β及TNF-α mRNA表达。结论:有氧运动通过抑制心肌梗死后NF-κB及其转录系统的激活,减少心肌组织炎症反应的过度放大,抑制交感神经过度再生,从而改善心肌梗死后交感神经重构。
关键词:有氧运动;心肌梗死;神经重构;nuclear factor-κB;炎症
中图分类号:G804.21文献标识码:A文章编号:1006-2076(2015)04-0068-06
心肌梗死(myocardial infarction, MI)是威胁人类健康的主要疾病之一,恶性室性心律失常及心源性猝死是MI患者恢复期致死、致残的重要原因。一系列研究证实MI后恶性室性心律失常高发与异常心脏交感神经重构导致的心脏自主神经失衡有关,同时MI后交感神经重构也可直接或间接地影响电重构及心肌重构,引起心电不稳定或心功能恶化[1-2]。目前认为心脏交感神经重构是MI后心脏病理生理发展的重要环节,探索MI后交感神经重构的具体机制将为降低恶性心律失常以及猝死发生率、改善远期预后提供新的治疗方向。
近年来的研究显示,心脏局部或全身炎症反应均可上调心肌中神经生长因子(nerve growth factor,NGF)表达,从而促进心脏交感神经再生。炎症反应作为神经再生的基础环节,在MI后交感神经重构的过程中发挥着关键作用,因此,抗炎有望成为改善MI后交感神经重构、降低心律失常发生率的有效手段。
有氧运动作为心血管疾病重要的康复手段逐渐成为运动医学领域研究的热点,MI早期适宜强度的有氧运动可通过逆转心肌重构[3]、增加每搏输出量及射血分数[4]、改善左室收缩功能[5]等发挥心脏保护作用。新近研究证实有氧运动具有显著的抗炎作用[6],Chen等[7]研究发现,有氧运动可显著改善MI大鼠左心室交感神经分布,降低交感神经再生,但其机制尚不明确。因此,本研究拟在MI大鼠模型基础上,以核转录因子-κB(nuclear factor-κB,NF-κB)及其下游炎症因子为研究主线,深入探讨有氧运动对炎症调控因子NF-κB及心脏交感神经再生的影响,从有氧运动抑制炎症反应角度探讨运动对MI后心脏交感神经重构的干预作用,从分子水平阐明运动防治MI后心源性猝死的可能机制。
1材料与方法
1.1实验材料
健康成年雄性SD大鼠45只,体重250~280 g(购自山东大学实验动物中心)。Trizol试剂盒购于美国Invitrogen公司;Real-time RT-PCR试剂盒购于大连宝生物工程公司,引物由上海生工生物工程公司设计并合成;细胞核蛋白及细胞浆蛋白抽提试剂盒购于碧云天生物技术研究所;抗酪氨酸羟化酶(tyrosine hydroxylase, TH)抗体购于美国Millipore公司,辣根过氧化物酶标记兔抗绵羊二抗购于美国PKL公司,抗NF-κB p65抗体购于美国BD公司,抗IκBα抗体及抗Histone 3抗体购于美国Cell Signaling公司,抗GAPDH抗体购于北京康为世纪生物科技有限公司。其余试剂均为国产分析纯。
1.2实验方法
1.2.1心肌梗死模型制备
SD大鼠随机分为假手术组(Sham组)、心肌梗死组(MI组)以及心肌梗死+有氧运动组(MI+AE组),每组15只。利用结扎冠状动脉左前降支的方法制备大鼠心肌梗死的模型[8],所有动物用10% 水合氯醛溶液0.3 mL /100 g腹腔注射麻醉,气管插管,小动物呼吸机通气。于大鼠胸骨左侧3-4肋间开胸,结扎左前降支,以心电图aVL导联ST段抬高0.2mV作为手术成功标志,随后逐层关胸。Sham组大鼠作为对照,只开胸穿线,不结扎。
MI+AE组大鼠手术恢复1周后参照Kemi OJ[9]的运动方案进行训练:动物跑台运动速度为15 m/min,大鼠进行适应运动10 min;将跑台运动速度增至20 m/min,持续运动50 min。大鼠运动60 min/d,5次/周,共计4周。Sham组及MI组正常笼内喂养不运动。[JP]
1.2.2心肌标本处理
实验大鼠4周有氧运动干预后,所有成活大鼠再次麻醉并迅速取下心脏,经主动脉灌注生理盐水清除残存血液。选取梗死灶周(梗死苍白区边缘3 mm内)及非梗死左室游离壁(梗死苍白区边缘2 cm以外)心肌,Sham组取相应部分心肌组织,分别存放于中性甲醛及液氮中保存。
1.2.3TH阳性神经纤维密度测定
取甲醛固定后的心肌组织,石蜡包埋,切片,片厚4 μm,常规脱蜡、高压修复后加一抗4°C过夜,二抗37 ℃孵育30 min,DAB显色,复染后封片。参照Cao等[10]的方法,选取神经分布较为密集的区域对神经纤维密度进行分析:采用Image Pro Plus5图像处理分析软件定量分析TH阳性神经纤维在所选区域中所占面积(以μm2/mm2表示),取其平均值作为阳性神经纤维密度值。
1.2.4胞核中NF-κB p65及胞质中IκBα蛋白表达
取100 g心肌,切成细小碎片,根据碧云天生产细胞核蛋白及细胞浆蛋白抽提说明书分别提取心肌组织中胞核及胞浆蛋白。用12%分离胶、5%浓缩胶100 V电泳60 min,160 V恒压电转60 min,5%脱脂奶粉封闭2 h后加一抗,4°C过夜,辣根过氧化物酶标记的二抗室温孵育1 h。ECL发光液显影,Image J 图像处理软件,以内参GAPDH或Histone 3蛋白条带光密度值为标准,计算目的蛋白相对表达量。
1.2.5IL-1β及TNF-α mRNA表达
根据Trizol试剂盒提供的方法提取心肌组织总mRNA。按照real-time RT-PCR试剂盒的方法进行逆转录及扩增。引物序列:IL-1β,上游 5-AGT GGC AAT GAA AAT GAC CTG-3,下游 5-CAC AAC GAC TGA CAA GAC CTG-3;TNF-α,上游5-CTG CCT CAG CCT CTT CTC TTT-3,下游 5-CAC TTG CGG GTT TGC TAC TAC-3;GAPDH,上游5-ACA GCA ACA GGG TGG TGG AC-3,下游5-TTT GAG GGT GCA GCG AAC TT-3。GAPDH作为内参,采用2-△△CT法计算目的基因相对表达量。
1.2.6统计学分析
所有数据均采用SPSS17.0统计软件进行统计学分析。计量数据以均数±标准差([AKx-]± s)表示,多组间比较采用单因素方差分析,然后采用最小显著差异法进行组间两两比较。P<0.05为统计学差异具有显著性。[JP]
2结果
2.1心肌中TH阳性神经纤维分布及表达
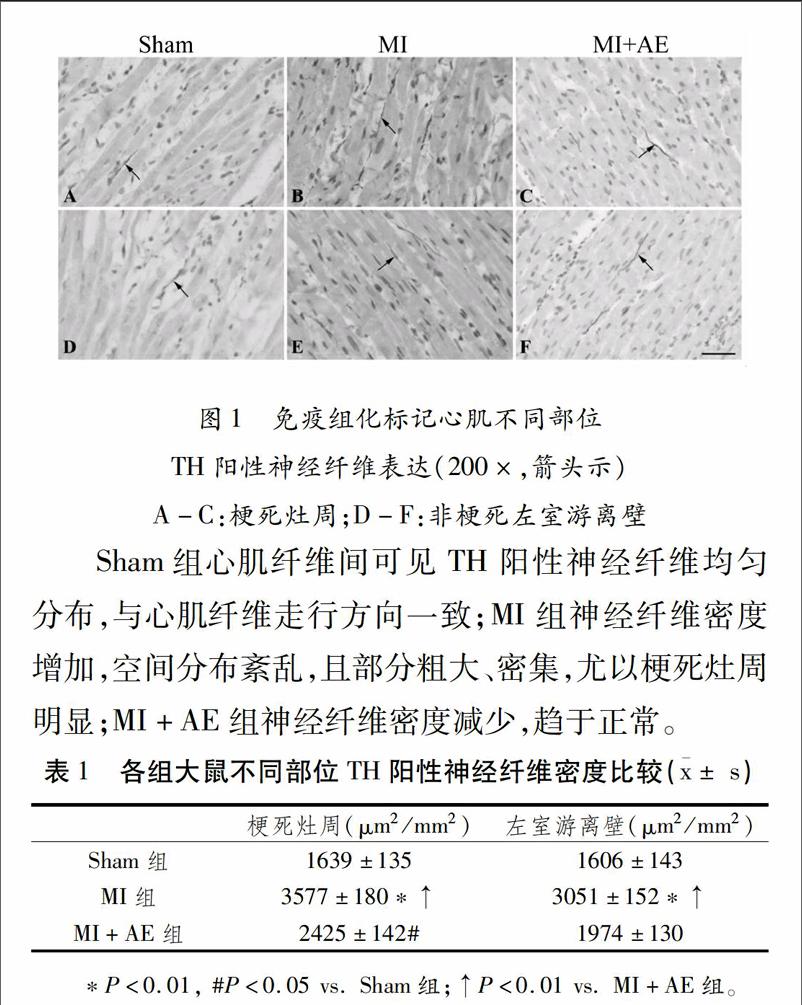
如图1所示,Sham组TH阳性神经纤维在心肌组织中均匀分布,沿心肌纤维纵行分布;MI组TH阳性神经纤维密度明显增加,尤以梗死灶周显著。神经纤维空间分布紊乱,形态异常,在小血管周围以及梗死灶周可见粗大、密集的TH阳性神经,部分聚集成束,偶可相互交错呈网状;与单纯MI组相比,MI+AE组梗死灶周及左室游离壁TH阳性神经纤维密度明显降低,形态更趋于正常。各组神经纤维密度统计详见表1
[TP4Q25.TIF,BP][TS(][HT5"K][JZ(]图1免疫组化标记心肌不同部位TH阳性神经纤维表达(200×,箭头示)
A-C:梗死灶周;D-F:非梗死左室游离壁[JZ)]
Sham组心肌纤维间可见TH阳性神经纤维均匀分布,与心肌纤维走行方向一致;MI组神经纤维密度增加,空间分布紊乱,且部分粗大、密集,尤以梗死灶周明显;MI+AE组神经纤维密度减少,趋于正常。
表1各组大鼠不同部位TH阳性神经纤维密度比较([AKx-]± s)
[BG(][BHDFG1*2,WK8,WK11,WKW][]梗死灶周(μm2/mm2)[]左室游离壁(μm2/mm2)
[BHD]Sham组[]1639±135[]1606±143
MI组[]3577±180*↑[]3051±152*↑
[BH]MI+AE组[]2425±142#[]1974±130[BG)F][HTK]*P<0.01, #P<0.05 vs. Sham组;↑P<0.01 vs. MI+AE组。
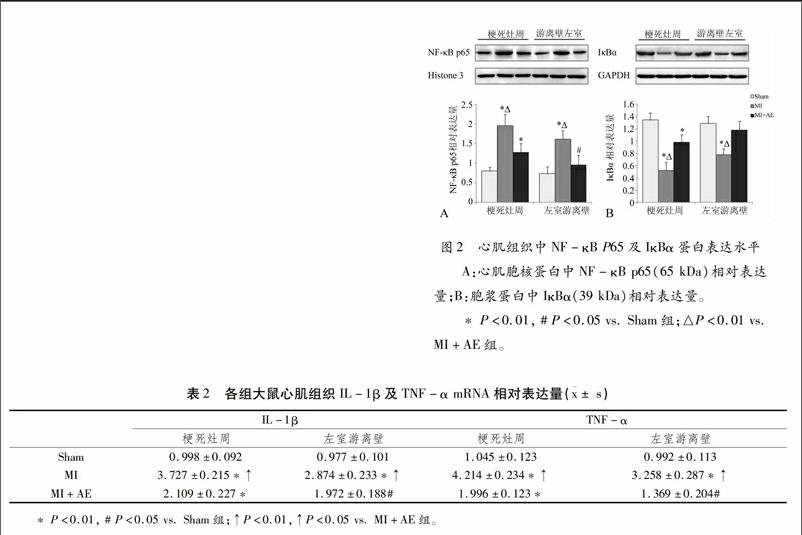
2.2心肌细胞核中NF-κB p65及细胞浆中 IκBα蛋白表达
如图2所示,MI后NF-κB通路明显激活。与Sham组相比,MI组梗死灶周和左室游离壁心肌细胞核蛋白中NF-κB p65蛋白表达量明显增加(梗死灶周:0.798±0.101 vs. 1.957±0.283,P<0.01;游离壁:0.727±0.183 vs. 1.614±0.215,P<0.01);此外,各部位心肌细胞浆蛋白中IκBα蛋白水平降低(梗死灶周:1.345±0.123 vs. 0.522±0.134,P<0.01;游离壁:1.287±0.113 vs. 0.779±0.097,P<0.01)。而有氧运动干预后,可显著抑制NF-κB通路激活,表现为心肌细胞核蛋白中NF-κB P65蛋白降低(梗死灶周:1.957±0.283 vs. 1.274±0.227,P<0.01;左室游离壁:1.614±0.215 vs. 0.956±0.238,P<0.01),且心肌细胞浆蛋白中IκBα蛋白水平明显增加(梗死灶周:0.522±0.134 vs. 0.981±0.123,P<0.01;左室游离壁:0.779±0.097 vs. 1.177±0.144,P<0.01)。2.3炎症因子IL-1β及TNF-α mRNA的表达
与NF-κB表达变化趋势一致,MI后梗死灶周和左室游离壁IL-1β及TNF-α相应mRNA表达水平
显著上调;有氧运动干预后可有效抑制NF-κB通路激活,其下游IL-1β及TNF-α转录水平明显被抑制,相应mRNA表达水平下调(表2)。
[TP4Q26.TIF,BP][TS(][HT5"K][JZ]图2心肌组织中NF-κB P65及IκBα蛋白表达水平
A:心肌胞核蛋白中NF-κB p65(65 kDa)相对表达量;B:胞浆蛋白中IκBα(39 kDa)相对表达量。
* P<0.01, # P<0.05 vs. Sham组;△P<0.01 vs. MI+AE组。[FL)0]
表2各组大鼠心肌组织IL-1β及TNF-α mRNA相对表达量([AKx-]± s)
[BG(][BHDFG3,WK10,WKW][][ZB(][BHDG1*2,WK24,WKW]IL-1β[]TNF-α
[BHDG1*2,WK12。3,WKW]梗死灶周[]左室游离壁[]梗死灶周[]左室游离壁[ZB)]
[BHDG1*2,WK10,WK12。3,WKW]Sham[]0.998±0.092[]0.977±0.101[]1.045±0.123[]0.992±0.113
MI[]3.727±0.215*↑[]2.874±0.233*↑[]4.214±0.234*↑[]3.258±0.287*↑
[BH]MI+AE[]2.109±0.227*[]1.972±0.188#[]1.996±0.123*[]1.369±0.204#[BG)F][HTK]
* P<0.01, # P<0.05 vs. Sham组;↑P<0.01,↑P<0.05 vs. MI+AE组。
3讨论
业已证实,MI后心脏交感神经纤维存在着变性、坏死、再生、重构的动态演变过程,心脏交感神经重构是交感神经对损伤刺激的再生和过度再生反应[11]。同其他组织再生一样,MI后神经再生是机体对组织损伤后的修复反应,适度的交感神经再生可在一定程度上改善MI后左心室结构、增加冠脉血供、维持血流动力学稳定[12-13],但异常的交感神经重构可改变心脏自主神经平衡,改变局部心肌细胞的自律性、传导性和不应期,加重MI后心脏电生理紊乱[14]。此外,心脏交感神经重构可改变离子通道各亚基编码基因表达[15]、加速心肌细胞凋亡及心肌纤维化[16],直接或间接影响电重构、心肌重构,三者相互叠加,成为触发MI后致死性心律失常及心源性猝死的关键因素。因此,对MI后心脏交感神经再生的调控被认为是预防或降低MI后致死性心律失常发生的有效手段。在本实验中,我们的研究从形态学上证实MI后存在交感神经重构现象,表现为TH阳性神经纤维密度明显增加,且空间分布紊乱,可见粗大、密集的TH阳性神经纤维,部分聚集成束,偶可相互交错呈网状,尤以梗死灶周为著,这一结果与既往研究结果一致[2,10]。而与单纯MI组相比,MI+AE组梗死灶周及左室游离壁心肌组织的TH阳性神经纤维密度显著减少,左室游离壁的神经纤维形态及分布更趋于正常。
MI后心脏交感神经再生是一个复杂的病理生理过程,其具体机制尚不明确。新近研究发现交感神经再生与炎症反应密切相关,MI后交感神经再生主要发生在炎症细胞和炎症因子等大量聚集的梗死灶周,炎症细胞及炎症因子通过多种途径上调NGF的表达,从而促进交感神经轴突延伸、生长[17-18]。多项研究证实,MI后心肌交感神经密度在时间-空间的动态表达变化与心肌局部炎症反应程度呈正相关[18-20]。Wang等[21]的研究更进一步证实,MI后NF-κB通路激活所诱导的炎症级联反应是交感神经再生的关键环节。NF-κB作为关键性的转录因子,在正常生理状态下,与其抑制因子IκB蛋白家族成员结合并形成复合体,以无活性形式存在于胞浆中;当细胞受到多种细胞外信号刺激后,NF-κB与IκB解离并由胞质移位至胞核,与DNA上的启动子区域相应靶基因位点结合,从而启动一系列免疫和炎症反应相关基因的转录,上调多种炎症因子表达,触发炎症级联反应[22]。因此,越来越多的学者认为NF-κB将成为炎症相关性疾病治疗的关键性靶点。在本实验中,我们利用Western blot方法检测心肌细胞核蛋白中NF-κB p65蛋白及胞浆蛋白中IκBα蛋白含量变化以明确NF-κB活化状态,结果显示MI组心肌细胞中NF-κB p65蛋白较Sham组显著增加,而IκBα蛋白含量降低,其下游IL-1β、TNF-α等炎症因子相应mRNA表达上调,尤以心肌梗死灶周为著。重要的是,在梗死灶周同样可以观察到显著的交感神经重构现象,这与Wang等的报道是一致的[21],我们的结果再次证实MI后NF-κB激活是促进心脏交感神经再生及重构的关键,也提示抑制NF-κB通路的激活可能成为改善MI后心脏交感神经重构的新靶点。
目前,关于有氧运动对心脏自主神经调控的研究已有诸多报道,Hautala等[23]在校正受试者年龄、训练时间等因素后发现,每周进行3次30 min预计最大心率60%至80%强度范围的有氧运动,并持续4周以上,可显著增强心脏迷走神经张力;Zoppini等[24]以2型糖尿病患者为研究对象,通过对心率变异性的动态监测发现有氧运动可降低LF/HF的比值,说明有氧运动可改善糖尿病患者心脏交感/迷走平衡,降低交感或增强迷走神经张力;Martinez等[25]通过对28名MI患者长达6个月的追踪观察发现,有氧运动可显著降低交感神经张力。但目前关于有氧运动调节心脏自主神经支配平衡的机制尚无分子学方面的合理解释。我们的研究发现,与单纯MI大鼠相比,MI+AE干预可有效抑制NF-κB通路激活,表现为心肌细胞核中NF-κB p65蛋白含量减少、胞浆中IκBα蛋白含量增加,且其下游IL-1β、TNF-α等炎症因子相应mRNA表达显著下调。Adamopoulos等[26]的研究表明,有氧运动可减少心衰患者外周血液中的炎症因子的表达,且有氧运动对心脏的保护作用与其抗炎作用密切相关。我们的研究更进一步证实,有氧运动通过 “抑制NF-κB激活—减轻炎症反应—下调NGF表达—抑制交感神经再生”这一连锁反应,改善MI后交感神经重构,这也可能是有氧运动降低交感神经张力、增强迷走神经张力的可能机制之一,这一结果也为有氧运动降低MI患者致死性心律失常发生率及猝死率提供了理论依据。
4结论
MI可导致NF-κB通路激活,介导炎症级联反应,通过上调心肌NGF表达促进心脏交感神经再生。有氧运动通过抑制MI后NF-κB通路激活,减轻MI后炎症连锁瀑布效应,从而改善心脏交感神经重构。
参考文献:
[1]Wang Y, Xuan YL, Hu HS,et al.Risk of ventricular arrhythmias after myocardial infarction with diabetes associated with sympathetic neural remodeling in rabbits [J].Cardiology,2012,121(1):1-9.
[2]Chen PS, Chen LS, Cao JM, Sharifi B, Karagueuzian HS, Fishbein MC. Sympathetic nerve sprouting, electrical remodeling and the mechanisms of sudden cardiac death [J]. Cardiovascular research,2001,50(2):409-16.
[3]Kraljevic J, Marinovic J, Pravdic D,et al.Aerobic interval training attenuates remodelling and mitochondrial dysfunction in the post-infarction failing rat heart [J]. Cardiovascular research,2013,99(1):55-64.
[4]Marshall KD, Muller BN, Krenz M,et al.Heart failure with preserved ejection fraction: chronic low-intensity interval exercise training preserves myocardial O2 balance and diastolic function [J]. J Appl Physiol (1985),2013,114(1):131-47.
[5]Giallauria F, Acampa W, Ricci F,et al.Exercise training early after acute myocardial infarction reduces stress-induced hypoperfusion and improves left ventricular function [J]. Eur J Nucl Med Mol Imaging,2013,40(3):315-24.
[6]Gleeson M, Bishop NC, Stensel DJ, Lindley MR, Mastana SS, Nimmo MA. The anti-inflammatory effects of exercise: mechanisms and implications for the prevention and treatment of disease [J]. Nature reviews Immunology,2011,11(9):607-15.
[7]Chen T, Cai MX, Li YY,et al.Aerobic exercise inhibits sympathetic nerve sprouting and restores beta-adrenergic receptor balance in rats with myocardial infarction [J]. PLoS One,2014,9(5):e97810.
[8]El-Helou V, Proulx C, Gosselin H,et al.Dexamethasone treatment of post-MI rats attenuates sympathetic innervation of the infarct region [J]. Journal of applied physiology,2008,104(1):150-6.
[9]Kemi OJ, Haram PM, Loennechen JP,et al.Moderate vs. high exercise intensity: differential effects on aerobic fitness, cardiomyocyte contractility, and endothelial function [J]. Cardiovascular research,2005,67(1):161-72.
[10]Cao JM, Fishbein MC, Han JB,et al.Relationship between regional cardiac hyperinnervation and ventricular arrhythmia [J]. Circulation,2000,101(16):1960-9.
[11]Vracko R, Thorning D, Frederickson RG. Fate of nerve fibers in necrotic, healing, and healed rat myocardium [J]. Laboratory investigation; a journal of technical methods and pathology,1990,63(4):490-501.
[12]Kiriazis H, Du XJ, Feng X,et al.Preserved left ventricular structure and function in mice with cardiac sympathetic hyperinnervation[J].American journal of physiology,2005,289(4):H1359-65.
[13]Fallen EL, Coates G, Nahmias C,et al.Recovery rates of regional sympathetic reinnervation and myocardial blood flow after acute myocardial infarction [J]. American heart journal,1999,137(5):863-9.
[14]Anderson KP. Sympathetic nervous system activity and ventricular tachyarrhythmias: recent advances [J]. Ann Noninvasive Electrocardiol,2003,8(1):75-89.
[15]Qu J, Robinson RB. Cardiac ion channel expression and regulation: the role of innervation [J]. Journal of molecular and cellular cardiology,2004,37(2):439-48.
[16]Singh K, Xiao L, Remondino A, Sawyer DB, Colucci WS. Adrenergic regulation of cardiac myocyte apoptosis [J]. Journal of cellular physiology,2001,189(3):257-65.
[17]Blasing H, Hendrix S, Paus R. Pro-inflammatory cytokines upregulate the skin immunoreactivity for NGF, NT-3, NT-4 and their receptor, p75NTR in vivo: a preliminary report [J]. Arch Dermatol Res,2005,296(12):580-4.
[18]Hasan W, Jama A, Donohue T,et al.Sympathetic hyperinnervation and inflammatory cell NGF synthesis following myocardial infarction in rats [J]. Brain Res,2006,1124(1):142-54.
[19]Wernli G, Hasan W, Bhattacherjee A, van Rooijen N, Smith PG. Macrophage depletion suppresses sympathetic hyperinnervation following myocardial infarction [J]. Basic research in cardiology,2009,104(6):681-93.
[20]Oh YS, Jong AY, Kim DT,et al.Spatial distribution of nerve sprouting after myocardial infarction in mice [J]. Heart Rhythm,2006,3(6):728-36.
[21]Wang Y, Suo F, Liu J,et al.Myocardial infarction induces sympathetic hyperinnervation via a nuclear factor-kappaB-dependent pathway in rabbit hearts [J]. Neuroscience letters,2013,535:128-33.
[22]Van der Heiden K, Cuhlmann S, Luong le A, Zakkar M, Evans PC. Role of nuclear factor kappaB in cardiovascular health and disease [J]. Clin Sci (Lond),2010,118(10):593-605.
[23]Hautala AJ, Makikallio TH, Kiviniemi A,et al.Cardiovascular autonomic function correlates with the response to aerobic training in healthy sedentary subjects [J]. American journal of physiology,2003,285(4):H1747-52.
[24]Zoppini G, Cacciatori V, Gemma ML,et al.Effect of moderate aerobic exercise on sympatho-vagal balance in Type 2 diabetic patients [J]. Diabet Med,2007,24(4):370-6.
[25]Martinez DG, Nicolau JC, Lage RL,et al.Effects of long-term exercise training on autonomic control in myocardial infarction patients [J]. Hypertension,2011,58(6):1049-56.
[26][JP+1]Adamopoulos S, Parissis J, Kroupis C,et al.Physical training reduces peripheral markers of inflammation in patients with chronic heart failure [J]. European heart journal,2001,22(9):791-7
