Dermatofibrosarcoma protuberans: from translocation to targeted therapy
2015-12-29JonathanNoujaimKhinThwayCyrilFisherRobinJones
Jonathan Noujaim, Khin Thway, Cyril Fisher, Robin L. Jones
Sarcoma Unit, Royal Marsden NHS Foundation Trust, London SW3 6JJ, UK
Dermatofibrosarcoma protuberans: from translocation to targeted therapy
Jonathan Noujaim, Khin Thway, Cyril Fisher, Robin L. Jones
Sarcoma Unit, Royal Marsden NHS Foundation Trust, London SW3 6JJ, UK
Dermatofbrosarcoma protuberans (DFSP), the most common dermal sarcoma, is a low-grade, slow growing fbroblastic malignant neoplasm that most frequently afects middle aged adults and is characterized by a high local recurrence rate and a low propensity for metastasis. Wide surgical resection or Mohs micrographic surgery (MMS) are the preferred approaches for localized disease, while radiation therapy is warranted for inoperable disease or for cases with positive margins where re-excision is not possible. DFSP is generally regarded as refractory to conventional chemotherapy. Treatment options for systemic disease were limited until the discovery of a unique translocation, t(17;22)(q22;q13) (COL1A1;PDGFB) found in a majority of cases. In recent years, imatinib, a PDGFβR, ABL and KIT inhibitor, has revolutionized systemic therapy in DFSP. In this review, we summarize the epidemiological, clinical, histological and genetic characteristics of DFSP and update the readers on its current management.
Dermatofbrosarcoma protuberans (DFSP); imatinib; Mohs micrographic surgery (MMS); translocation; targeted therapy
Introduction
Dermatofibrosarcoma protuberans (DFSP), although rare, is one of the most common dermal sarcomas1. It is a low-grade, slow growing fibroblastic malignant neoplasm most frequently affecting middle aged adults and characterized by a high local recurrence rate and a low propensity for metastasis. Clinically, DFSP consists initially of a slow growing violaceous, blue-red or brown indurated plaque confined to the dermis with subsequent nodules appearing at later stages. Histologically, it is represented by neoplastic spindle cells arranged in storiform or cartwheel paterns, difusely infltrating the dermis and subcutis. Nuclei generally show no atypia and the mitotic count is low. On immunohistochemical (IHC) studies, tumors typically show diffuse expression of CD34. Despite surgical resection, a substantial number of patients will recur and an unfortunate few will eventually develop metastatic disease. Compared to other sarcoma subtypes, DFSP is generally regarded as refractory to standard chemotherapy. A breakthrough in systemic treatment was made possible following the identification of a unique translocation t(17;22)(q22;q13) (COL1A1;PDGFB) in a majority of cases. This discovery subsequently led to the evaluation of imatinib, a PDGFβR, ABL and KIT inhibitor in the treatment of DFSP. In this review, we summarize the epidemiological, clinical, histological and genetic characteristics of DFSP and update the readers on its current management.
Epidemiology and clinical features
Te incidence of DFSP is estimated to be around 0.8-4.2 cases per million persons per year2. Due to better recognition and improvement in ancillary diagnostic modalities, the number of reported cases has increased steadily over the last decades. While DFSP most frequently affects middle age adults, it has been reported in adolescents as well as adults up to the 9th decade (age ranged 15 to over 90 years). There is a slight male predominance3,4. Pediatric cases of DFSP account for 6% of all diagnoses and require a high degree of suspicion5. A recent review of the literature shows that several pediatric cases havebeen reported so far; this might suggest that the number of infants with the condition might be larger than that estimated previously6. Clinical presentation of DFSP can be so diverse, particularly in children and adolescence that the differential diagnosis may not lead to malignancy. A biopsy is often the first step that can help to find the tumor as early as possible. Clearly, therefore, an accurate knowledge of the disease is the prerequisite for a wider recognition and appropriate treatment. The most commonly affected anatomical sites are the trunk (40%-50%), the proximal extremities (30%-40%), and the head and neck (10%-15%)3,7,8. Patients generally report a long history, ranging from months to years, of a slow growing indurated plaque. Lesions have a hard consistency and are fixed to the skin and may appear violaceous, blue-red or brown. They can vary considerably in size, ranging from 2 cm at early stages to 30 cm and larger in neglected cases, although size has not been shown to correlate with prognosis8,9. Te disease usually remains contained in the dermis and subcutis, although recurrent or long standing cases may invade deeper structures including the fascia, muscle, periosteum (particularly in cases afecting the scalp), and bone. At an early (‘preprotuberant’) stage, DFSP can be classifed into three different forms: a morphea-like form mimicking a scar, morphea, morphea-form basal cell carcinoma, or a dermatofibroma plaque; an atrophoderma-like form similar to atrophoderma or anetoderma; and fnally an angioma-like form resembling vascular malformations10. Eventually, one or multiple nodules may appear in the protuberant phase (Figure 1). Efforts should be made in excluding fibrosarcomatous transformation, which may occur de novo or upon recurrence, in patients reporting rapid enlargement of their neoplasm11-13. While the development of metastatic disease is rare (5%), rates of local recurrence are significant (20%-50%)7,8,14, with median time to local recurrence of around 32 months; longterm follow-up is therefore mandatory3. The lungs are the primary site for distal spread; although nodal involvement is reported uncommon4. Te global prognosis of DFSP is excellent, with reported 2- and 5-year survival rates of 97% and 92%, respectively15.
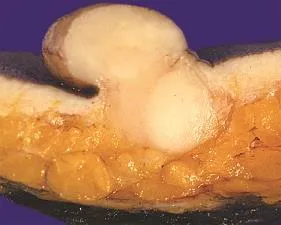
Figure 1 A large polypoid nodule is presented with a homogeneous frm yellowish white cut surface. The tumor can be seen to expand the entire dermis, and extends into the more superfcial subcutis.
Histology
On histopathological examination, DFSP is usually centered in the dermis and subcutis, and is an ill-defned, cellular neoplasm composed of storiform or ‘cartwheel’ distributions of bland, relatively monotonous spindle cells, with elongated nuclei with even chromatin, minimal cytological atypia and small amounts of fbrillary cytoplasm, within collagenous stroma (Figure 2A-C). The mitotic count is usually low. The tumor often infiltrates the subcutis in characteristic, linearly orientated strands that show a ‘honeycomb’ pattern. Immunohistochemically, tumors show diffuse and strong expression of CD34 (Figure 2D)16, but are negative for other markers such as cytokeratins, desmin, smooth muscle actin (SMA) and S100 protein. CD34 positivity supports a diagnosis of DFSP, but is not specifc as its expression is also found in a variety of spindle cell neoplasms in the histological differential diagnosis of DFSP, including some benign fbrohistiocytic lesions, solitary fbrous tumor and some undifferentiated spindle cell sarcomas. Furthermore, CD34 expression is also seen in a variety of neoplasms of other lineages, including vascular and hematopoietic tumors. Apolipoprotein D is also strongly expressed in DFSP and may be helpful in supporting its diagnosis and differentiating it from fibrous histiocytoma17.
While morphologically conventional DFSP accounts for more than 90% of cases, a number of histologic subtypes have been described. On occasions, there can be prominent myxoid stroma (myxoid DFSP) (Figure 2E)18,19, or rarely myoid or myofibroblastic differentiation, in which there are interspersed bundles of bland and SMA-positive myofbroblastic cells. Pigmented DFSP (also known as Bednar tumor) (Figure 2F)20,21is characterized by spindle cells in a storiform pattern in which there are scattered pigmented, dendritic melanocytic cells. It is more commonly found in black patients and behaves similarly to conventional DFSP. Atrophic DFSP is identifed by a depressed plaque with a reduced dermal thickness thereby displacing the subcutis closer to the epithelium22,23. Sclerosing DFSP shows less cellular foci, with homogenous collagen bundles, although other areas are morphologically identical to conventional DFSP24-26. Granular cell variant ofDFSP is rarely reported and consists of spindle cells mixed with a population of cells with abundant lysosomal granules, round eccentric nuclei, and prominent nucleoli27,28. In children, giant cell fbroblastoma (GCF) is considered a variant of DFSP and shares a common morphological, clinical and genetic background29,30. Areas of DFSP are seen in approximately 15% of GCF29. Areas of GCF are typically hypocellular, with bland spindle cells and interspersed tumor giant cells in patternless distributions within myxoid or collagenous stroma (Figure 2G). Finally, tumors that have undergone fibrosarcomatous transformation show a more loosely fascicular architecture (Figure 2H), rarely with cellular atypia, as well as more prominent mitotic figures, and often show loss of CD34 expression11,31. These changes are found in 10%-20% of cases, and comprise the most aggressive variant, associated with the highest risk of local recurrence and distant metastasis, and overall, are associated with a worse prognosis32,33.
Genetic background
Early cytogenetic studies initially documented the presence of supernumerary ring chromosomes in DFSP and subsequent fluorescence in situ hybridization (FISH) studies and comparative genomic hybridization (CGH) concluded that these rings derived from the centromere of chromosome 22 and interspersed sequences of chromosome 1734-37. Groundbreaking molecular work further revealed that these rearranged chromosomes contained sequences of α1 type I collagen preprotein gene (COL1A1;17q21) and platelet-derived growth factor β-chain gene (PDGFB;22q13)38. In approximately 90% of cases (and regardless of histological patern), tumors have the characteristic reciprocal translocation t(17;22)(q22;q13), with a majority in the form of supernumerary ring chromosomes. Te COL1A1 breakpoint is highly variable (spanning the exon 6 to exon 49 region) compared to the PDGFB breakpoint consistently found in intron 1. Te resultant gene fusion leads to constitutive upregulation of PDGFβ expression39. PDGF, a potent mitogen for mesenchymal cells, is involved in major signaling pathways including Ras-MAPK and PI3K-AKT-mTOR, and in stimulating cell growth, differentiation, and migration40,41. The fusion protein COL1A1-PDGFβ is processed to a mature PDGF-BB homodimer and activates the PDGFβ receptor (PDGFβR)leading to autocrine activation and subsequent tumorigenesis. It is highly probable that the acquisition of t(17;22)(q22;q13) is an early event in the clonal evolution of the disease because of the pre-clinical evidence demonstrating that transfection of the gene rearrangement is capable of transforming normal cells to neoplastic cells42,43.
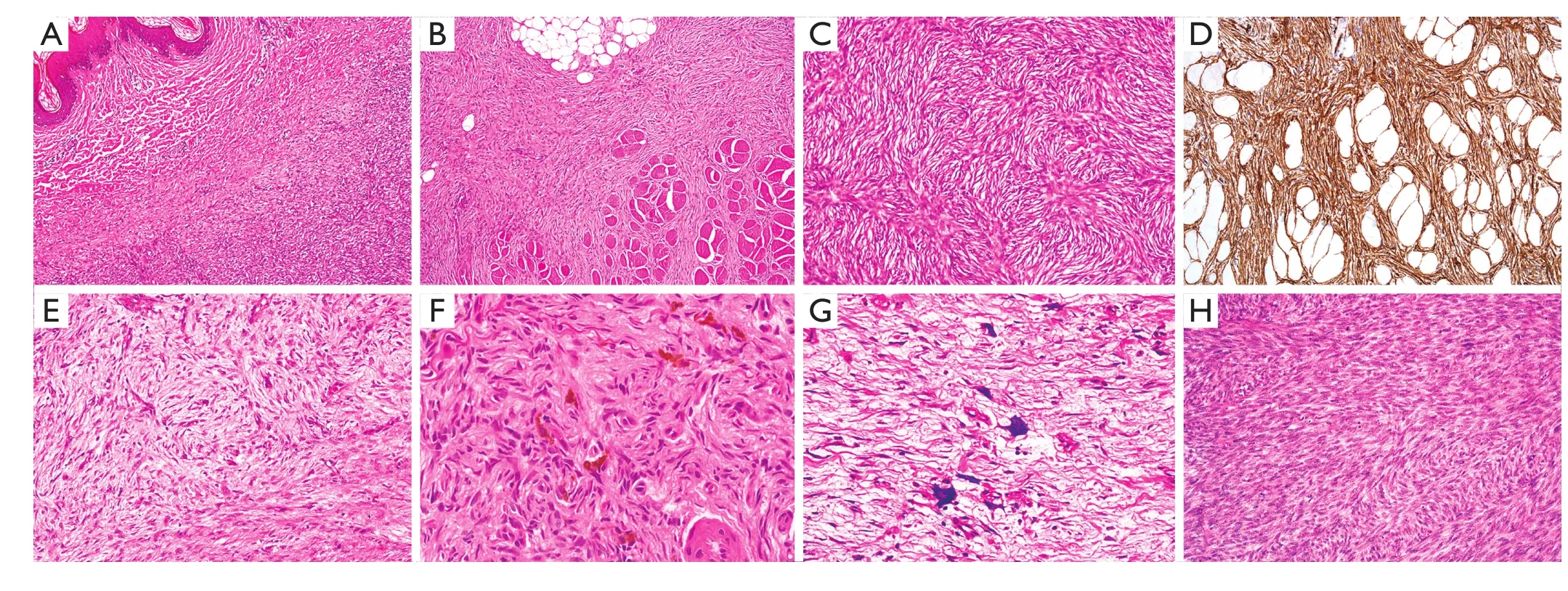
Figure 2 (A) Dermatofibrosarcoma protuberans (DFSP). Ill-defined cellular tumor is present within the superficial dermis, close to the squamous epithelium (top left of feld) (H&E staining, 20×). (B) Most DFSP infltrates the dermis and subcutis, but some are deeply infltrating. This example shows prominent infltration of skeletal muscle bundles (lower half of feld) (H&E staining, 20×). (C) DFSP typically comprises cellular distributions of bland spindle cells with elongated vesicular nuclei and small amounts of fbrillary cytoplasm, in a prominent storiform or ‘cartwheel’ pattern (H&E staining, 200×). (D) Immunohistochemically, DFSP characteristically shows diffuse and strong expression of CD34. This example also illustrates the linearly oriented tumor strands that infltrate the subcutaneous fat in a ‘honeycomb’ pattern (IHC staining, 100×). (E) Tumors may show focal or sometimes prominent myxoid change. As the characteristic storiform architecture is lost, there may be diffculty in establishing a diagnosis of DFSP (H&E staining, 100×). (F) Bednar tumor is characterized by spindle cells in a storiform pattern in which there are scattered pigmented, dendritic melanocytic cells (H&E staining, 400×). (G) In children, giant cell fbroblastoma (GCF) is considered a variant of DFSP, but GCF are typically hypocellular, with bland spindle cells and interspersed tumor giant cells in patternless distributions within myxoid or collagenous stroma (H&E staining, 400×). (H) In fbrosarcomatous transformation, the cells are present in more loosely fascicular distributions or ‘herringbone’ patterns, and often show more prominent mitotic activity (H&E staining, 100×).
The characteristic t(17;22) of DFSP can be detected in routine practice using FISH or multiplex reverse transcription polymerase chain reaction (RT-PCR) studies. FISH studies are easy, sensitive and only require PDGFB or COL1A1-PDGFB fusion probes whereas multiplex RT-PCR requires multiple COL1A1 primers because of the hypervariable breakpoint region44-46. The t(17;22) translocation is also identifiable in DFSP variants including pigmented DFSP, myxoid DFSP and fibrosarcomatous DFSP19. Fibrosarcomatous transformation can present de novo or result from long standing disease. Interestingly, genomic gains of COL1A1-PDGFβ were found in areas where DFSP evolved into fibrosarcomatous DFSP, once again providing evidence of its oncogenic properties47. However, other genomic events including microsatellite instability and p53 mutations are also involved in tumor progression in DFSP48. Beyond the oncological properties of the fusion gene COL1A1-PDGFβ, litle is known about the other mechanisms contributing to its tumorigenicity. The overexpression of p-STAT3, p-ERK and thrombospondin-1 were highlighted in IHC studies and may possibly contribute to its oncogenesis49,50. Activation of epidermal growth factor receptor (EGFR) and insulin receptor were also noted in a majority of clinical samples51. Furthermore, the role of miRNAs remains unexplored, but may play a pivotal role. Recently, down-regulation of miR-205 in DFSP was shown to lead to an overexpression of LRP-1 and possibly ERK phosphorylation thereby contributing to cellular proliferation52. Little is known about the mechanisms involved in COL1A1-PDGFβ negative DFSP. A case report described a DFSP patient lacking the characteristic COL1A1-PDGFβ, but instead presented a complex translocation between chromosomes 5 and 8, involving the CSPG2 gene at 5q14.3 and the PTK2B gene at 8p21.253. Clearly, more work is needed to fully understand the molecular background of DFSP, with a particular attention to t(17;22) negative DFSP.
Treatment
The current treatment options of DFSP were reviewed systematically by Rutkowski et al.54. In the following sections, we will highlight the important aspects on its management.
Surgery
The mainstay of management of localized disease is complete surgical resection7. Because of the locally invasive nature of DFSP, obtaining clear surgical margins with particular atention to the deep fascia is very important. When DFSP was initially approached with conservative margins, the reported recurrence rates were up to 60%4. By further removing the tumor en bloc with 2-5 cm margins, recurrence rates dropped around 6% but remained significant14,55-57. The underlying reason why DFSP tends to relapse despite wide surgical excision is explained by its tendency to invade outwards from the original focus of disease. At the time of surgery, these microscopic projections are not observable8. By further widening the excision in order to obtain more than 5 cm clear margins, the recurrence rates drop to almost 0%58,59. However, this comes at a cost of an increased risk of complication as well a sub-optimal cosmetic result. In recent years, outcomes following surgical resection were improved by the use of Mohs micrographic surgery (MMS). MMS is a technique that involves immediate examination of the microscopic margin60-64. The technique is repeated several times until the process is complete. Tis of course can enhance the cosmetic result and is important to consider in children and young adults. In addition, it can be particularly useful in lesions at challenging anatomic sites, where performing radical surgery can be difcult57,65,66. Lateral safety margins of 1-1.3 cm are sufcient67. Although some reviews suggest lower recurrence rates (around 1%) with MMS compared to conventional wide surgical resection (6.3%)56,57,62,63,65, conclusions cannot be drawn because of a lack of randomized trials. Despite this shortcoming, when available, MMS should be the privileged approach67. If unavailable, wide surgical resection with 3 cm margins is sufcient67,68.
Radiation therapy
Tere is no indication of adjuvant radiation therapy following an R0 resection. It may have a role following resection of a tumor with close or positive margins in which further surgery is not possible, especially when fibrosarcomatous transformation has occurred69. In addition, radiation may be employed as primary therapy in patients with unresectable disease70-72. The target volume should include the primary tumor volume in addition to a safety margin of 3-5 cm. The total recommended dose varies from 60 Gy for microscopic disease to 70 Gy for macroscopic disease in 2 Gy fractions67.
Imatinib therapy
An unfortunate minority of patients will subsequently metastasize during the course of their disease. DFSP is generally perceived as being resistant to conventional chemotherapy71,73. Tere is some anecdotal evidence of response to methotrexate, although the experience is limited74,75. A breakthrough in the management of metastatic patients was possible following the discovery of the translocation t(17;22)(q22;q13) (COL1A1;PDGFB), whereby efforts were directed towards targeting PDGFR. Imatinib is an inhibitor of PDGFβR, ABL and KIT, and blocks PDGF signaling, interfering with phosphorylation of the receptor tyrosine kinase. Preclinical studies showed that imatinib had in vitro and in vivo activity via the induction of apoptosis (rather than reducing proliferation)76,77. Imatinib activity was also observed in tumors expressing low levels of PGDFRB. Recent transcriptional profiling of imatinib-treated tumor samples was suggestive of a possible enhanced susceptibility of IM-treated tumor cells to an antigen specific, HLA-restricted immune recognition78. In 2002, two clinical reports documented the activity of this drug in patients with inoperable or metastatic disease79,80. A patient with locally advanced DFSP achieved a response to imatinib enabling sufficient downstaging for surgery to be performed. On histopathological examination, there was a pathological complete response. Another patient, with DFSP with fibrosarcomatous change, did not respond to imatinib79. Te other report documented a good response in a patient with a tumor harbouring the t(17;22) translocation80. The clinical activity of imatinib was subsequently described in several other case reports81,82.
The Imatinib Target Exploration Consortium Study B2225 proceeded to show that eight patients with locally advanced disease harboring the t(17;22) translocation responded to 800 mg of imatinib (4 obtaining complete responses). No response was documented in a metastatic patient lacking the translocation83. Te results of this study subsequently led to Food and Drug Administration (FDA) approval of imatinib for DFSP. Similar results were also observed for DFSP patients enrolled in a phase II trial studying imatinib in tumors with imatinib sensitive tyrosine kinases84. A combined analysis of the SWOG and EORTC phase II trials (prematurely closed because of slow recruitment) in patients with locally advanced and metastatic DFSP has been published85. In the EORTC trial patients were required to have the PDGFβ rearrangement to be eligible to enter the trial, and patients could undergo surgery at 14 weeks if this was deemed feasible. Patients entered into the EORTC trial (n=16) were treated with imatinib 400 mg twice per day while those in the SWOG trial (n=8) received 400 mg once per day. A total of 24 patients were included in the analysis with a median follow-up of 2.6 years; 11 (46%) achieved a Response Evaluation Criteria in Solid Tumors (RECIST) partial response and four experienced progressive disease. Te median time to progression was 1.7 years. Tis trial confrmed the previous reported activity of imatinib in DFSP, but also suggested that 400 mg once a day may be sufcient.
Neo-adjuvant imatinib
Imatinib was also studied in a neo-adjuvant setting with the aim to improve on surgical outcomes. A multicenter phase II trial of neoadjuvant imatinib 600 mg once a day for a 2-month period prior to surgical resection reported a clinical response rate in 9 of 25 patients (36%)86; the lower response rate here can be explained by the short duration of imatinib treatment. A further multicenter phase II trial evaluated neoadjuvant imatinib 600 mg once a day for a minimum of 6 weeks51. Thereafter if the patient had at least stable disease, imatinib was continued at the discretion of the investigator until the planned surgery. The reported response rate was 57.1%, similar to previous imatinib trials. Te median treatment duration was 3.1 months and the median follow-up was 6.4 years. Only one patient developed secondary resistance to imatinib. Follow-up did not support smaller surgical margins following successful response to imatinib. These studies show once again that lower doses of imatinib are sufficient and well tolerated in treatment of DFSP. However, it is difcult to comment on the impact of neoadjuvant imatinib therapy on surgical margins and recurrence rates. The initial French study proceeded with wide excision but did not document any follow-up86while the Ugurel et al.51trial proceeded to narrow to intermediate excision (<2 cm). Therefore, direct comparison is impossible. The routine use of neo-adjuvant imatinib for operable DFSP cannot be recommended and should remain investigational. However, its use is warranted in borderline resectable cases. Under these circumstances, neo-adjuvant imatinib should be continued until a maximal response is documented before proceeding to surgery.
Imatinib resistance and translocation negative DFSP
Patients lacking the t(17;22) translocation do not usually respond to imatinib51,83,86. Furthermore, the pigmented-variant DFSP seems to be unresponsive to imatinib despite cases presenting the translocation, although larger cohorts will be needed to validate these fndings51,85. To our knowledge, there arecurrently no trials investigating novel agents designed specifcally for DFSP. Treatment options available for translocation negative or imatinib-resistant DFSP are limited. Single case reports have suggested that other tyrosine kinase inhibitors such as sunitinib51and sorafenib73,87may have some activity in tumors resistant to imatinib. Further investigations are required to evaluate the full spectrum of activity of these drugs. DFSP with fbrosarcomatous transformation can respond to imatinib; however, resistance appears rapidly78,88,89. Because fbrosarcomatous transformation carries a worse prognosis, patients with inoperable or metastatic disease should be treated with conventional anthracycline-based schedules following imatinib failure90.
Efforts are now directed towards understanding imatinib resistance in DFSP. Whole genome sequencing in a patient with DFSP who subsequently developed imatinib resistance did not reveal point mutations in the PDGFB gene91. Instead, novel mutations were found in genes implicated in various signaling pathways including NK-κB. Therefore, it is possible that resistance is secondary to other mechanisms rather than the acquisition of new mutations in the COL1A1-PDGFB fusion gene. Recently, homozygous deletion of CDKN2A and CDKN2B were identified in pre-clinical imatinib-resistant models92. CDK4/6 inhibitors were able to inhibit cellular proliferation in vitro and warrant further investigation.
Conclusion
DFSP is a rare soft tissue sarcoma subtype with an infiltrative growth patern and low rate of metastatic disease. Te mainstay of management of localized disease is surgical resection, and MMS or wide surgical resection is the current accepted approaches. Radiation can be administered following resection with close or positive margins (if re-excision is impossible) and can be primary therapy for patients with inoperable disease. Conventional chemotherapy has limited activity in this disease. In the subset of patients with inoperable or metastatic disease, imatinib therapy is warranted following confirmation of t(17;22)(q22;q13) (COL1A1;PDGFB). A starting dose of 400 mg once a day is sufcient and beter tolerated. For imatinibresistant patients, options are limited and include sorafenib and sunitinib. Enrollment in clinical trials is strongly encouraged. Eforts should now be directed towards beter understanding the biology of DFSP in order to identify novel targets.
Acknowledgements
We acknowledge support from the NIHR Royal Marsden/ICR Biomedical Research Center.
Conflict of interest statement
No potential conficts of interest are disclosed.
1. Fletcher CDM BJ, Hogendoorn PCW, Mertens F. WHO Classifcation of Tumours of Sof Tissue and Bone. Pathology and Genetics of Tumours of Sof Tissue and Bone. IARC Press 2013.
2. Criscione VD, Weinstock MA. Descriptive epidemiology of dermatofbrosarcoma protuberans in the United States, 1973 to 2002. J Am Acad Dermatol 2007;56:968-973.
3. Bowne WB, Antonescu CR, Leung DH, Katz SC, Hawkins WG, Woodruf JM, et al. Dermatofbrosarcoma protuberans: A clinicopathologic analysis of patients treated and followed at a single institution. Cancer 2000;88:2711-2720.
4. Rutgers EJ, Kroon BB, Albus Luter CE, Gortzak E. Dermatofbrosarcoma protuberans: treatment and prognosis. Eur J Surg Oncol 1992;18:241-248.
5. Valdivielso-Ramos M, Hernanz JM. Dermatofbrosarcoma protuberans in childhood. Actas Dermosifliogr 2012;103:863-873.
6. Manganoni AM, Pavoni L, Gualdi G, Marocolo D, Chiudinelli M, Sereni E, et al. Dermatofbrosarcoma protuberans in an adolescent: a case report and review of the literature. J Pediatr Hematol Oncol 2013;35:383-387.
7. Chang CK, Jacobs IA, Salti GI. Outcomes of surgery for dermatofbrosarcoma protuberans. Eur J Surg Oncol 2004;30:341-345.
8. Gloster HM. Dermatofbrosarcoma protuberans. J Am Acad Dermatol 1996;35:355-374; quiz 375.
9. Khatri VP, Galante JM, Bold RJ, Schneider PD, Ramsamooj R, Goodnight JE Jr. Dermatofbrosarcoma protuberans: reappraisal of wide local excision and impact of inadequate initial treatment. Ann Surg Oncol 2003;10:1118-1122.
10. Martin L, Piete F, Blanc P, Mortier L, Avril MF, Delaunay MM, et al. Clinical variants of the preprotuberant stage of dermatofibrosarcoma protuberans. Br J Dermatol 2005;153:932-936.
11. Wrotnowski U, Cooper PH, Shmookler BM. Fibrosarcomatous change in dermatofbrosarcoma protuberans. Am J Surg Pathol 1988;12:287-293.
12. Ding J, Hashimoto H, Enjoji M. Dermatofbrosarcoma protuberans with fbrosarcomatous areas. A clinicopathologic study of nine cases and a comparison with allied tumors. Cancer 1989;64:721-729.
13. Connelly JH, Evans HL. Dermatofbrosarcoma protuberans. A clinicopathologic review with emphasis on fbrosarcomatous areas. Am J Surg Pathol 1992;16:921-925.
14. Fiore M, Miceli R, Mussi C, Lo Vullo S, Mariani L, Lozza L, et al. Dermatofbrosarcoma protuberans treated at a single institution: a surgical disease with a high cure rate. J Clin Oncol 2005;23:7669-7675.
15. Corey RM, Swet K, Ward WG. Epidemiology and survivorship of sof tissue sarcomas in adults: a national cancer database report. Cancer Med 2014;3:1404-1415.
16. Weiss SW, Nickolof BJ. CD-34 is expressed by a distinctive cell population in peripheral nerve, nerve sheath tumors, and related lesions. Am J Surg Pathol 1993;17:1039-1045.
17. West RB, Harvell J, Linn SC, Liu CL, Prapong W, Hernandez-Boussard T, et al. Apo D in sof tissue tumors: a novel marker for dermatofbrosarcoma protuberans. Am J Surg Pathol 2004;28:1063-1069.
18. Reimann JD, Fletcher CD. Myxoid dermatofbrosarcoma protuberans: a rare variant analyzed in a series of 23 cases. Am J Surg Pathol 2007;31:1371-1377.
19. Mentzel T, Schärer L, Kazakov D, Michal M. Myxoid dermatofbrosarcoma protuberans: clinicopathologic, immunohistochemical, and molecular analysis of eight cases. Am J Dermatopathol 2007;29:443-448.
20. Fletcher CD, Teaker JM, Flanagan A, Krausz T. Pigmented dermatofbrosarcoma protuberans (Bednar tumour): melanocytic colonization or neuroectodermal diferentiation? A clinicopathological and immunohistochemical study. Histopathology 1988;13:631-643.
21. Yagi Y, Ueda K, Maruyama S, Noborio R. Bednar tumor: a report of two cases. J Dermatol 2004;31:484-487.
22. Martin L, Combemale P, Dupin M, Chouvet B, Kanitakis J, Bouyssou Gauthier ML, et al. Te atrophic variant of dermatofbrosarcoma protuberans in childhood: a report of six cases. Br J Dermatol 1998;139:719-725.
23. Zelger BW, Ofner D. Atrophic variants of dermatofbroma and dermatofibrosarcoma protuberans. Histopathology 1995;26:519-527.
24. Diaz-Cascajo C, Weyers W, Borghi S. Sclerosing dermatofibrosarcoma protuberans. J Cutan Pathol 1998;25:440-444.
25. Hatori H. Nodular sclerotic change in dermatofbrosarcoma protuberans: a potential diagnostic problem. Br J Dermatol 2003;148:357-360.
26. Sabater-Marco V, Pérez-Vallés A, Berzal-Cantalejo F, Rodriguez-Serna M, Martinez-Diaz F, Martorell-Cebollada M. Sclerosing dermatofbrosarcoma protuberans (DFSP): an unusual variant with focus on the histopathologic diferential diagnosis. Int J Dermatol 2006;45:59-62.
27. Banerjee SS, Harris M, Eyden BP, Hamid BN. Granular cell variant of dermatofbrosarcoma protuberans. Histopathology 1990;17:375-378.
28. Maire G, Pédeutour F, Coindre J-M. COL1A1-PDGFB gene fusion demonstrates a common histogenetic origin for dermatofbrosarcoma protuberans and its granular cell variant. Am J Surg Pathol 2002;26:932-937.
29. Terrier-Lacombe MJ, Guillou L, Maire G, Terrier P, Vince DR, de Saint Aubain Somerhausen N, et al. Dermatofbrosarcoma protuberans, giant cell fbroblastoma, and hybrid lesions in children: clinicopathologic comparative analysis of 28 cases with molecular data--a study from the French Federation of Cancer Centers Sarcoma Group. Am J Surg Pathol 2003;27:27-39.
30. Jha P, Moosavi C, Fanburg Smith J. Giant cell fbroblastoma: an update and addition of 86 new cases from the Armed Forces Institute of Pathology, in honor of Dr. Franz M. Enzinger. Ann Diagn Pathol 2007;11:81-88.
31. Goldblum JR. CD34 positivity in fbrosarcomas which arise in dermatofbrosarcoma protuberans. Arch Pathol Lab Med 1995;119:238-241.
32. Hoesly PM, Lowe GC, Lohse CM, Brewer JD, Lehman JS. Prognostic impact of fbrosarcomatous transformation in dermatofbrosarcoma protuberans: a cohort study. J Am Acad Dermatol 2015;72:419-425.
33. Liang CA, Jambusaria-Pahlajani A, Karia PS, Elenitsas R, Zhang PD, Schmults CD. A systematic review of outcome data for dermatofbrosarcoma protuberans with and without fbrosarcomatous change. J Am Acad Dermatol 2014;71:781-786.
34. Bridge JA, Nef JR, Sandberg AA. Cytogenetic analysis of dermatofbrosarcoma protuberans. Cancer Genet Cytogenet 1990;49:199-202.
35. Mandahl N, Heim S, Willén H, Rydholm A, Mitelman F. Supernumerary ring chromosome as the sole cytogenetic abnormality in a dermatofbrosarcoma protuberans. Cancer Genet Cytogenet 1990;49:273-275.
36. Pedeutour F, Coindre JM, Sozzi G, Nicolo G, Leroux A, Toma S, et al. Supernumerary ring chromosomes containing chromosome 17 sequences. A specifc feature of dermatofbrosarcoma protuberans? Cancer Genet Cytogenet 1994;76:1-9.
37. Pedeutour F, Simon MP, Minoleti F, Sozzi G, Pieroti MA, Hecht F, et al. Ring 22 chromosomes in dermatofbrosarcoma protuberans are low-level amplifers of chromosome 17 and 22 sequences. Cancer Res 1995;55:2400-2403.
38. Simon MP, Pedeutour F, Sirvent N, Grosgeorge J, Minoleti F, Coindre JM, et al. Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofbrosarcoma protuberans and giant-cell fbroblastoma. Nat Genet 1997;15:95-98.
39. Takahira T, Oda Y, Tamiya S, Higaki K, Yamamoto H, Kobayashi C, et al. Detection of COL1A1-PDGFB fusion transcripts andPDGFB/PDGFRB mRNA expression in dermatofbrosarcoma protuberans. Mod Pathol 2007;20:668-675.
40. Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev 1999;79:1283-1316.
41. Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev 2008;22:1276-1312.
42. Greco A, Fuseti L, Villa R, Sozzi G, Minoleti F, Mauri P, et al. Transforming activity of the chimeric sequence formed by the fusion of collagen gene COL1A1 and the platelet derived growth factor b-chain gene in dermatofbrosarcoma protuberans. Oncogene 1998;17:1313-1319.
43. Shimizu A, O’Brien KP, Sjablom T, Pietras K, Buchdunger E, Collins VP, et al. Te dermatofbrosarcoma protuberans-associated collagen type Ialpha1/platelet-derived growth factor (PDGF) B-chain fusion gene generates a transforming protein that is processed to functional PDGF-BB. Cancer Res 1999;59:3719-3723.
44. Kerob D, Pedeutour F, Leboeuf C, Verola O, de Kerviler E, Servant J, et al. Value of cytogenetic analysis in the treatment of dermatofibrosarcoma protuberans. J Clin Oncol 2008;26:1757-1759.
45. Salgado R, Llombart B, M Pujol R, Fernández-Serra A, Sanmartín O, Toll A, et al. Molecular diagnosis of dermatofbrosarcoma protuberans: a comparison between reverse transcriptasepolymerase chain reaction and fuorescence in situ hybridization methodologies. Genes Chromosomes Cancer 2011;50:510-517.
46. Patel KU, Szabo SS, Hernandez VS, Prieto VG, Abruzzo LV, Lazar AJ, et al. Dermatofbrosarcoma protuberans COL1A1-PDGFB fusion is identifed in virtually all dermatofbrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fuorescence in situ hybridization assays. Hum Pathol 2008;39:184-193.
47. Abbot JJ, Erickson-Johnson M, Wang X, Nascimento AG, Oliveira AM. Gains of COL1A1-PDGFB genomic copies occur in fbrosarcomatous transformation of dermatofbrosarcoma protuberans. Mod Pathol 2006;19:1512-1518.
48. Takahira T, Oda Y, Tamiya S, Yamamoto H, Kawaguchi K, Kobayashi C, et al. Microsatellite instability and p53 mutation associated with tumor progression in dermatofbrosarcoma protuberans. Hum Pathol 2004;35:240-245.
49. Lin N, Urabe K, Moroi Y, Uchi H, Nakahara T, Dainichi T, et al. Overexpression of phosphorylated-STAT3 and phosphorylated-ERK protein in dermatofbrosarcoma protuberans. Eur J Dermatol 2006;16:262-265.
50. Maekawa T, Jinnin M, Ohtsuki M, Ihn H. Te expression levels of thrombospondin-1 in dermatofbroma and dermatofbrosarcoma protuberans. Eur J Dermatol 2011;21:534-538.
51. Ugurel S, Mentzel T, Utikal J, Helmbold P, Mohr P, Pföhler C, et al. Neoadjuvant imatinib in advanced primary or locally recurrent dermatofbrosarcoma protuberans: a multicenter phase II DeCOG trial with long-term follow-up. Clin Cancer Res 2014;20:499-510.
52. Kajihara I, Jinnin M, Harada M, Makino K, Honda N, Makino T, et al. miR-205 down-regulation promotes proliferation of dermatofbrosarcoma protuberans tumor cells by regulating LRP-1 and ERK phosphorylation. Arch Dermatol Res 2014;306:367-374.
53. Bianchini L, Maire G, Guillot B, Joujoux JM, Follana P, Simon MP, et al. Complex t(5;8) involving the CSPG2 and PTK2B genes in a case of dermatofbrosarcoma protuberans without the COL1A1-PDGFB fusion. Virchows Arch 2008;452:689-696.
54. Rutkowski P, Debiec-Rychter M. Current treatment options for dermatofbrosarcoma protuberans. Expert Rev Anticancer Ter 2015;15:901-909.
55. DuBay D, Cimmino V, Lowe L, Johnson T, Sondak V. Low recurrence rate afer surgery for dermatofbrosarcoma protuberans: a multidisciplinary approach from a single institution. Cancer 2004;100:1008-1016.
56. Paradisi A, Abeni D, Rusciani A, Cigna E, Wolter M, Scuderi N, et al. Dermatofbrosarcoma protuberans: wide local excision vs. Mohs micrographic surgery. Cancer Treat Rev 2008;34:728-736.
57. Foroozan M, Sei JF, Amini M, Beauchet A, Saiag P. Efcacy of Mohs micrographic surgery for the treatment of dermatofbrosarcoma protuberans: systematic review. Arch Dermatol 2012;148:1055-1063.
58. Arnaud EJ, Perrault M, Revol M, Servant JM, Banzet P. Surgical treatment of dermatofbrosarcoma protuberans. Plast Reconstr Surg 1997;100:884-895.
59. Lemm D, Mügge LO, Mentzel T, Höfen K. Current treatment options in dermatofbrosarcoma protuberans. J Cancer Res Clin Oncol 2009;135:653-665.
60. Wacker J, Khan Durani B, Hartschuh W. Modifed Mohs micrographic surgery in the therapy of dermatofbrosarcoma protuberans: analysis of 22 patients. Ann Surg Oncol 2004;11:438-444.
61. Meguerditchian AN, Wang J, Lema B, Kraybill W, Zeitouni N, Kane J. Wide excision or Mohs micrographic surgery for the treatment of primary dermatofbrosarcoma protuberans. Am J Clin Oncol 2010;33:300-303.
62. Serra-Guillén C, Llombart B, Nagore E, Guillén C, Requena C, Traves V, et al. Mohs micrographic surgery in dermatofbrosarcoma protuberans allows tumour clearance with smaller margins and greater preservation of healthy tissue compared with conventional surgery: a study of 74 primary cases. Br J Dermatol 2015;172:1303-1307.
63. Snow SN, Gordon EM, Larson PO, Bagheri MM, Bentz ML, Sable DB. Dermatofbrosarcoma protuberans: a report on 29 patients treated by Mohs micrographic surgery with long-term follow-up and review of the literature. Cancer 2004;101:28-38.
64. Loghdey MS, Varma S, Rajpara SM, Al-Rawi H, Perks G, Perkins W. Mohs micrographic surgery for dermatofbrosarcoma protuberans (DFSP): a single-centre series of 76 patients treated by frozensection Mohs micrographic surgery with a review of the literature. J Plast Reconstr Aesthet Surg 2014;67:1315-1321.
65. Ratner D, Tomas CO, Johnson TM, Sondak VK, Hamilton TA, Nelson BR, et al. Mohs micrographic surgery for the treatment of dermatofbrosarcoma protuberans. Results of a multiinstitutional series with an analysis of the extent of microscopic spread. J Am Acad Dermatol 1997;37:600-613.
66. Nouri K, Lodha R, Jimenez G, Robins P. Mohs micrographic surgery for dermatofbrosarcoma protuberans: University of Miami and NYU experience. Dermatol Surg 2002;28:1060-1064; discussion 1064.
67. Saiag P, Grob JJ, Lebbe C, Malvehy J, Del Marmol V, Pehamberger H, et al. Diagnosis and treatment of dermatofbrosarcoma protuberans. European consensus-based interdisciplinary guideline. Eur J Cancer 2015. [Epub ahead of print].
68. Bichakjian CK, Olencki T, Alam M, Andersen JS, Berg D, Bowen GM, et al. Dermatofbrosarcoma protuberans, version 1.2014. J Natl Compr Canc Netw 2014;12:863-868.
69. Dagan R, Morris C, Zlotecki R, Scarborough M, Mendenhall W. Radiotherapy in the treatment of dermatofbrosarcoma protuberans. Am J Clin Oncol 2005;28:537-539.
70. Haas RL, Keus RB, Lofus BM, Rutgers EJ, van Coevorden F, Bartelink H. Te role of radiotherapy in the local management of dermatofbrosarcoma protuberans. Sof Tissue Tumours Working Group. Eur J Cancer 1997;33:1055-1060.
71. Ballo MT, Zagars GK, Pisters P, Pollack A. Te role of radiation therapy in the management of dermatofbrosarcoma protuberans. Int J Radiat Oncol Biol Phys 1998;40:823-827.
72. Castle KO, Guadagnolo BA, Tsai CJ, Feig BW, Zagars GK. Dermatofbrosarcoma protuberans: long-term outcomes of 53 patients treated with conservative surgery and radiation therapy. Int J Radiat Oncol Biol Phys 2013;86:585-590.
73. Fields RC, Hameed M, Qin LX, Moraco N, Jia X, Maki RG, et al. Dermatofbrosarcoma protuberans (DFSP): predictors of recurrence and the use of systemic therapy. Ann Surg Oncol 2011;18:328-336.
74. Mendoza CB, Gerwig WH, Watne AL. Dermatofbrosarcoma protuberans with metastases treated with methotrexate. Am J Surg 1970;120:119-121.
75. Ng A, Nishikawa H, Lander A, Grundy R. Chemosensitivity in pediatric dermatofbrosarcoma protuberans. J Pediatr Hematol Oncol 2005;27:100-102.
76. Greco A, Roccato E, Miranda C, Cleris L, Formelli F, Pieroti MA. Growth-inhibitory efect of STI571 on cells transformed by the COL1A1/PDGFB rearrangement. Int J Cancer 2001;92:354-360.
77. Sjöblom T, Shimizu A, O’Brien KP, Pietras K, Dal Cin P, Buchdunger E, et al. Growth inhibition of dermatofbrosarcoma protuberans tumors by the platelet-derived growth factor receptor antagonist STI571 through induction of apoptosis. Cancer Res 2001;61:5778-5783.
78. Stacchioti S, Pantaleo MA, Negri T, Astolf A, Tazzari M, Dagrada GP, et al. Efcacy and biological activity of imatinib in metastatic dermatifbrosarcoma protuberans (DFSP). Clin Cancer Res 2015. [Epub ahead of print].
79. Maki RG, Awan R, Dixon RH, Jhanwar S, Antonescu CR. Diferential sensitivity to imatinib of 2 patients with metastatic sarcoma arising from dermatofbrosarcoma protuberans. Int J Cancer 2002;100:623-626.
80. Rubin BP, Schuetze SM, Eary JF, Norwood TH, Mirza S, Conrad EU, et al. Molecular targeting of platelet-derived growth factor B by imatinib mesylate in a patient with metastatic dermatofbrosarcoma protuberans. J Clin Oncol 2002;20:3586-3591.
81. Mizutani K, Tamada Y, Hara K, Tsuzuki T, Saeki H, Tamaki K, et al. Imatinib mesylate inhibits the growth of metastatic lung lesions in a patient with dermatofbrosarcoma protuberans. Br J Dermatol 2004;151:235-237.
82. Price VE, Fletcher JA, Zielenska M, Cole W, Viero S, Manson DE, et al. Imatinib mesylate: an atractive alternative in young children with large, surgically challenging dermatofbrosarcoma protuberans. Pediatr Blood Cancer 2005;44:511-555.
83. McArthur GA, Demetri GD, van Oosterom A, Heinrich MC, Debiec-Rychter M, Corless CL, et al. Molecular and clinical analysis of locally advanced dermatofbrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol 2005;23:866-873.
84. Heinrich MC, Joensuu H, Demetri GD, Corless CL, Apperley J, Fletcher JA, et al. Phase II, open-label study evaluating the activity of imatinib in treating life-threatening malignancies known to be associated with imatinib-sensitive tyrosine kinases. Clin Cancer Res 2008;14:2717-2725.
85. Rutkowski P, Van Glabbeke M, Rankin C, Ruka W, Rubin B, Debiec Rychter M, et al. Imatinib mesylate in advanced dermatofbrosarcoma protuberans: pooled analysis of two phase II clinical trials. J Clin Oncol 2010;28:1772-1779.
86. Kérob D, Porcher R, Vérola O, Dalle S, Maubec E, Aubin F, et al. Imatinib mesylate as a preoperative therapy in dermatofbrosarcoma: results of a multicenter phase II study on 25 patients. Clin Cancer Res 2010;16:3288-3295.
87. Kamar FG, Kairouz VF, Sabri AN. Dermatofbrosarcoma protuberans (DFSP) successfully treated with sorafenib: case report. Clin Sarcoma Res 2013;3:5.
88. Gronchi A, Stacchioti S, Pedeutour F, Collini P, Tamborini E, Morosi C, et al. Response to imatinib mesylate (IM) infbrosarcoma (FS) arising in dermatofbrosarcoma protuberans (DFSP). J Clin Oncol (Meeting Abstracts) 2008;26:10593.
89. Rutkowski P, Dębiec Rychter M, Nowecki Z, Michej W, Symonides M, Ptaszynski K, et al. Treatment of advanced dermatofbrosarcoma protuberans with imatinib mesylate with or without surgical resection. J Eur Acad Dermatol Venereol 2011;25:264-270.
90. Labropoulos SV, Fletcher JA, Oliveira AM, Papadopoulos S, Razis ED. Sustained complete remission of metastatic dermatofbrosarcoma protuberans with imatinib mesylate. Anticancer Drugs 2005;16:461-466.
91. Hong JY, Liu X, Mao M, Li M, Choi DI, Kang SW, et al. Genetic aberrations in imatinib-resistant dermatofbrosarcoma protuberans revealed by whole genome sequencing. PLoS One 2013;8:e69752.
92. Eilers G, Czaplinski J, Mayeda M, Bahri N, Tao D, Zhu M, et al. CDKN2A/p16 loss implicates CDK4 as a therapeutic target in imatinib-resistant dermatofbrosarcoma protuberans. Mol Cancer Ter 2015;14:1346-1353.
Cite this article as: Noujaim J, Thway K, Fisher C, Jones RL. Dermatofibrosarcoma protuberans: from translocation to targeted therapy. Cancer Biol Med 2015;12:375-384. doi: 10.7497/j.issn.2095-3941.2015.0067
Robin L. Jones
E-mail: robin.jones@rmh.nhs.uk
Received August 23, 2015; accepted September 15, 2015.
Available at www.cancerbiomed.org
Copyright © 2015 by Cancer Biology & Medicine
杂志排行

Cancer Biology & Medicine的其它文章
- Mechanistic basis and clinical relevance of the role of transforming growth factor-β in cancer
- PI3K/Akt/mTOR inhibitors in breast cancer
- Advances in drug delivery system for platinum agents based combination therapy
- Upgrading the definition of early gastric cancer: better staging means more appropriate treatment
- Synergistic suppression of the PI3K inhibitor CAL-101 with bortezomib on mantle cell lymphoma growth
- Pathological clavicular fracture as first presentation of renal cell carcinoma: a case report and literature review