改性石墨烯纳米材料气敏特性的理论研究
2015-12-24
改性石墨烯纳米材料气敏特性的理论研究
唐亚楠,申梓刚,陈卫光,李成刚,潘立军
郑州师范学院量子材料研究中心,物理与电子工程学院,郑州450044
摘要:利用基于密度泛函理论的第一性原理计算方法,研究单个CO和O2气体分子在多种金属原子修饰的石墨烯表面的吸附作用.结果表明,空位缺陷结构的石墨烯能够显著提高金属原子的稳定性,失去部分电荷的金属原子有助于调控气体分子的吸附特性.对比发现,单个金属Al和Mo原子掺杂的石墨烯体系对O2分子具有极高的灵敏性和选择性.通过不同气体分子的吸附能够调控石墨烯体系的电子结构和磁性.研究结果测试了不同金属原子修饰石墨烯表面的反应活性,为设计新型金属-石墨烯功能器件提供参考.
关键词:表面与界面物理学;密度泛函理论;石墨烯;气敏性能;电子结构;磁性;功能器件
Received: 2015-04-01; Accepted: 2015-04-15
Foundation: National Natural Science Foundation of China(U1404109) ; Science Fund of Educational Department of Henan Province(14B140019)
以半导体为气敏材料的传感器具有体积小、灵敏度高、响应快和成本低的特点,被广泛用于工业、环保、气体泄漏、气象监测及自动控制等领域.相比传统的半导体材料,石墨烯具有机械特性好[1]、化学性能稳定[2]、导电性和热传导性好等优点[3],极高的比表面积[4]有助提高其作为催化剂的使用效率.因易于集成和功能化,石墨烯基气敏器件具有广阔的应用前景.气敏传感器是通过被检测气体与传感器表面发生反应,引起体系某种性质发生变化(电导或电阻等)来工作的.气体分子作为电子给体或受体吸附在石墨烯表面会引起体系电子结构的变化.
碳原子间sp2杂化作用使石墨烯具有较强的化学惰性,使完整结构的石墨烯与吸附气体间的相互作用较弱[5],降低了石墨烯对被测气体分子的灵敏性.通常情况下,引入原子吸附或掺杂能调控石墨烯的表面结构和反应活性[6].研究发现,金属原子掺杂的石墨烯体系稳定性高,能有效分散催化剂,提高使用效率[7].掺杂不同的单个金属原子有利于调控石墨烯体系的电子结构、磁性和催化活性[8-9].Ambrosi等[10]验证了石墨烯材料中可存在金属杂质原子.Wang等[11]研制出不同金属原子(Pt、Co和In)掺杂的石墨烯功能材料.金属原子掺杂的石墨烯体系能提高对HCN、NO2和NH3等气体分子的探测灵敏性[12-14].然而O2和CO作为最普遍的气体分子,对因这两种气体吸附而引起改性石墨烯体系的电子结构的研究却较少,尤其缺乏对多种金属原子修饰的石墨烯体系气敏特性的系统研究.
本研究基于密度泛函理论的第一性原理计算方法,研究不同金属原子修饰石墨烯体系的稳定性、体系电子结构的变化规律和其调控气体分子(O2和CO)吸附特性的能力.通过比较吸附能的大小,测试单个金属原子对气体分子的灵敏性,分析气体分子和改性石墨烯衬底间的作用机理,为设计石墨烯基气敏器件提供理论参考.
1 计算参数与模型
采用基于密度泛函理论的第一性原理软件包VASP(Vienna ab-inito simulation package)[15-16]进行数值计算,交换相关泛函使用广义梯度近似(generalized gradient approximation,GGA)下的PBE(Perdew-Burke-Ernzerhof)[17]来处理,而离子实通过投影缀加波赝势(projector augmented wave,PAW)[18]表示,平面波基组截断能为450 eV.布里渊区积分的Monkhorst-Pack网格点为5×5×1[19].超原胞选取单层4×4石墨烯,平面取x和y方向,z方向垂直于平面,设真空层厚1.5 nm.考虑自旋极化效应,抹平参数σ= 0.2 eV,自洽迭代和离子弛豫的收敛标准分别设为能量收敛至1.0×10-5eV,原子力收敛至0.2 eV/nm.采用Bader电荷分析技术[20],确定体系中原子的电荷数,通过比较反应前后原子电荷数的变化来描述电荷转移量.
如图1(a),a和b分别表示基矢方向,完整结构石墨烯具有3个高对称吸附位,分别是桥位(B,位于C—C键的中点)、顶位(T,位于碳原子的正上方向)和空位(H,位于六方蜂巢格子的正中心).单个金属原子(metal atom,MA)替代石墨烯中的一个碳原子形成金属原子掺杂石墨烯(MA-graphene)的稳定结构,本研究以Fe原子掺杂石墨烯的构型为例,主要考虑气体分子在掺杂金属原子顶部的吸附情况,通过对比体系总能量大小来探寻最稳定构型.

图1 金属Fe原子掺杂石墨烯体系的俯视和侧视图及其电荷密度图(等高线均以5.6×108C/m3画出)Fig.1(Color online) Top and side views of the Fe embedded-graphene and the valence charge density plots(The contour lines in plots are drawn at about 5.6×108C/m3)
体系的吸附能定义为

其中,EA为单个原子(或分子)的能量(包括Al、Fe、Cu、Zn、Ni、Co、Mo、CO和O2) ; EB为石墨烯体系的能量(包括完整结构、空位缺陷的石墨烯和MA-graphene衬底) ; EA/B为原子(或分子)吸附石墨烯体系的总能量.吸附能越大意味着相应结构的稳定性越高.
金属原子在空位缺陷处的形成能定义为

其中,EAM-graphene为金属原子掺杂石墨烯体系的能量; EC和EMA分别为单个碳原子和金属原子的能量; Epri为完整石墨烯体系的能量.
2 结果与讨论
2.1单个金属原子在石墨烯衬底上的稳定构型
优化后的石墨烯晶格常数a = b = 0.248 nm; C—C键长为0.143 nm,这与文献[21]的实验结果(a =b = 0.246 nm,C—C键长为0.142 nm)十分接近.比较3个高对称位的吸附能,单个金属原子在完整结构石墨烯衬底的吸附能分别为: H位的Fe原子(1.19 eV)、Co原子(1.65 eV)、Ni原子(1.42 eV)和Al原子(1.03 eV),B位的Cu原子(0.21 eV)和Mo原子(0.09 eV),Zn原子不吸附.比较吸附能可知,不同金属原子吸附在高对称位表现出稳定性的差异.观察单个金属原子的扩散运动,发现吸附能较大的Al、Fe、Co和Ni原子在石墨烯表面的反应势垒较小(0.21~0.49 eV),意味着这些金属原子在石墨烯表面容易通过移动形成团簇结构.因此,完整结构的石墨烯并不能起到分散和提高催化剂稳定性的作用.
通常采用单空位缺陷结构来提高石墨烯对金属原子的束缚作用.Fe原子掺杂石墨烯形成的稳定结构如图1(a)和(b)所示.相比完整结构的石墨烯,掺杂了金属原子的石墨烯,其金属原子在空位缺陷处的吸附能较大(表1).由于金属原子的半径都大于碳原子的,这些掺杂的原子将从石墨烯表面突出出来.通过观察图1(c),可分析掺杂金属原子和紧邻碳原子的相互作用.提供电荷的Fe原子引起周围碳原子电荷的重新分布,在Fe—C界面电荷积聚度越高,相互作用越强,说明碳原子与掺杂原子形成的共价键提高了金属原子的稳定性.采用Bader分析计算电荷转移量,掺杂的Mo和Al原子向石墨烯体系提供了较多电荷.表1列举了7种金属原子掺杂石墨烯体系的吸附能Eads,金属原子和紧邻碳原子间距离LM-C,金属原子失去的电荷量ΔC,吸附高度hD和形成能Eform.观察单个金属原子掺杂石墨烯的形成能可知,相比Cu、Zn和Al金属原子的Eform,掺杂Fe、Co、Ni和Mo原子的Eform较小(<1.0 eV),说明这些金属原子易束缚在空位缺陷处成为石墨烯的金属杂质,这与文献[10]实验结果一致.掺杂的金属原子因失去电荷而显正电性,并将作为石墨烯表面的活性位来调控气体分子的吸附特性.

表1 金属原子掺杂石墨烯体系的Eads、LM-C、
2.2 CO和O2在MA-graphene上的吸附特性
通过研究金属原子掺杂石墨烯体系稳定结构,测试单个气体分子在MA-graphene体系的稳定吸附构型.图2为CO和O2吸附在Fe-graphene体系的稳定结构.平行吸附的O2与Fe原子形成2个Fe—O键,键长0.187 nm,Eads= 2.01 eV.吸附的CO分子中C原子垂直指向Fe-graphene表面,其Fe—C键长为0.189 nm,Eads=1.10 eV.
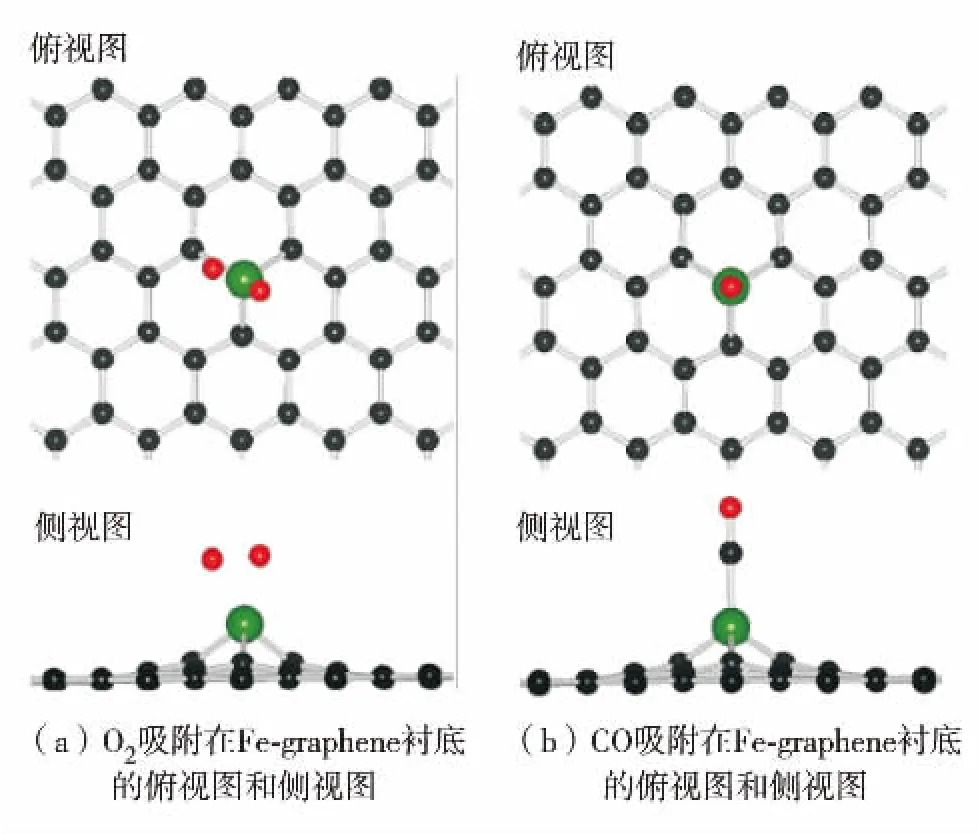
图2 O2和CO分别吸附在Fe-graphene衬底的俯视和侧视图Fig.2(Color online) Top and side views of the stable configurations of O2adsorbed Fe-graphene and CO adsorbed Fe-graphene
表2比较了不同气体分子吸附在MA-graphene体系的稳定性.其中,dMA-O2/CO为金属原子与气体分子间的距离; Eads为O2/CO分子吸附能; dO-O/C-O为
气体分子键长;ΔC1为吸附气体获得电荷数;μ为吸附体系总磁矩.与其他MA-graphene衬底相比,单个O2(2.57 eV)和CO(0.89 eV)在Mo-graphene表面吸附的能量差值最大.吸附的CO分子获得较少的电荷,稳定性较低.相比之下,O2分子在MA-graphene衬底上更易获得电荷,且稳定性较高.此外,吸附的O2获得电荷数增多会导致O O键伸长.由于O2的吸附能大于CO的,若以混合CO/O2作为反应气体,MA-graphene表面更易被O2占据,有助于进行氧化反应.

表2 O2和CO吸附在MA-graphene衬底的稳定性参数Table 2 The stable configuration data of O2adsorbed MA-graphene and CO adsorbed MA-graphene
2.3气体吸附在MA-graphene体系的电子结构
完整结构的石墨烯体系具有零带隙,而单空位缺陷的石墨烯体系因存在不饱和电子态,会在费米能级处出现尖峰[22].采用态密度(density of state,DOS)图分析因掺杂金属原子引起石墨烯体系电子结构的变化,如图3.相比空位缺陷的石墨烯体系,掺杂的金属原子向空位缺陷处的碳原子悬挂键提供转移电荷,使MA-graphene体系总态密度(total density of state,TDOS)分布发生明显变化.Co、Zn 和Cu掺杂的石墨烯体系表现出半金属特性,Fe、Ni和Mo掺杂的石墨烯体系表现出半导体特性,说明这些掺杂的金属原子能有效调控石墨烯体系的电子结构.在费米能级附近,Co、Fe、Ni和Mo原子中d轨道波峰与体系总电子态部分重叠,说明掺杂的金属原子与石墨烯衬底间相互作用很强.

图3 金属原子掺杂石墨烯体系的态密度图Fig.3 DOS plots of MA-graphene systems
单个CO和O2气体吸附引起MA-graphene体系电子结构发生变化.如图4(a),以Fe-graphene体系为例,在费米能级附近,吸附的O2使体系杂质态明显增多,呈半导体特性.Fe-3d轨道、O2-2π*、5σ轨道与体系总电子态分布发生部分重叠,说明O2与Fe-graphene的相互作用很强.Bader分析技术算出从Fe-graphene体系转移到O2上的电荷为0.71 e,转移电荷主要集中在O2-2π*轨道.吸附的O2分子改变了体系中未成对电子数,使Fe-graphene体系的自旋上和自旋下轨道分布不对称且显磁性(μ = 2.0 μB,其中μB=(9.274 015 4±0.000 003 1)× 10-24A·m2).图4(b)为CO吸附的Fe-graphene体系.在费米能级附近,Fe-3d轨道与CO-2π*和5σ轨道间形成了较强的杂化作用.Bader分析技术算出从Fe-graphene转移到CO上的电荷量为0.25e,这些电荷占据CO-2π*轨道导致C—O键伸长到0.116 nm(表2).在费米能级附近,相比吸附气体前的Fe-3d态波峰分布,气体分子吸附后的Fe-3d态峰值降低且分布变宽,意味着Fe-3d态的部分电子转移到吸附的气体分子上,因此,掺杂的Fe原子提供的电荷增强了气体分子与石墨烯衬底的相互作用.与吸附的O2相比,CO吸附并未引起Fe-graphene体系的磁性变化.同样,在Zn-graphene衬底,CO的吸附并未引起体系的磁性变化,吸附的O2增强了体系的磁性(μ= 2.0 μB).此外,与Fegraphene和Zn-graphene衬底相比,CO吸附的Mographene体系具有磁性(μ=2.0μB),O2吸附的石墨烯体系不显磁性.因此,在MA-graphene衬底上,通过吸附不同的气体分子可有效调制改性石墨烯体系的电子结构和磁性变化.
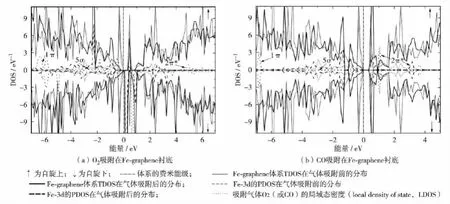
图4 单个O2和CO吸附在Fe-graphene衬底的态密度图Fig.4 Spin-resolved DOS plots of O2adsorbed Fe-graphene and CO adsorbed Fe-graphene
掺杂的金属原子提供的电荷部分转移到吸附的气体分子,部分转移到空位缺陷处的碳原子悬挂键,说明金属原子起到调控体系表面结构和稳定吸附气体的作用.由于吸附能的大小决定探测气体的灵敏性和选择性,在MA-graphene衬底上,O2获得电荷量(0.46e~1.02e)和吸附能(0.94~2.57 eV)的变化都较大,而CO获得电荷量(0.20e~0.33e)和吸附能的变化范围(0.34~1.10 eV)较小,说明O2是敏感气体分子,易受环境变化影响.若将混合的CO/O2作为反应气体,MA-graphene表面更易被获取电荷能力更强的O2占据.此外,在Al-graphene和Mo-graphene表面,O2吸附能远大于CO的能量.在Ni-、Co-、Fe-、Zn-和Cu-graphene衬底中,O2和CO吸附能相当,有利促进气体分子间的相互作用.
结语
本研究采用基于密度泛函理论的第一性原理方法计算CO和O2分子在多种金属原子修饰石墨烯表面的吸附特性.研究发现,金属原子掺杂的石墨烯体系稳定性高,掺杂的金属原子失去电荷后显正电性,石墨烯体系表现出半导体或半金属特性.与CO分子相比,吸附的O2分子更易获得电荷,转移电荷的数量能够促进改性石墨烯与气体分子的相互作用.在所研究的金属掺杂的石墨烯体系中,Al和Mo原子掺杂的石墨烯体系对吸附的O2更具灵敏度和选择性.对于Ni、Co、Cu、Fe和Zn原子掺杂的石墨烯衬底,CO和O2的吸附能相差较小,有利于进行气体分子间的相互作用.因此,金属原子修饰的石墨烯纳米材料将在气体传感器、自旋电子器件和催化电极等方面具有广泛的应用前景.
引文:唐亚楠,申梓刚,陈卫光,等.改性石墨烯纳米材料气敏特性的理论研究[J].深圳大学学报理工版,2015,32(4) : 365-370.
参考文献/References:
[1]Lee C,Wei X,Kysar J W,et al.Measurement of the elastic properties and intrinsic strength of monolayer graphene[J].Science,2008,321(5887) : 385-388.
[2]Biswas C,Lee Y H.Graphene versus carbon nanotubes in electronic devices[J].Advanced Functional Materials,2011,21(20) : 3806-3826.
[3]Novoselov K,Geim A,Morozov S,et al.Two-dimensional gas of massless Dirac fermions in graphene[J].Nature,2005,438(7065) : 197-200.
[4]Meyer J.Carbon sheets an atom thick give rise to graphene dreams[J].Science,2009,324(5929) : 875-877.
[5]Zou Y,Li F,Zhu Z,et al.An ab initio study on gas sensing properties of graphene and Si-doped graphene [J].European Physical Journal B,2011,81(4) : 475-479.
[6]Tang Qing,Zhou Zhen,Chen Zhongfang.Graphenerelated nanomaterials: tuning properties by functionalization[J].Nanoscale,2013,5(11) : 4541-4583.
[7]Tang Ya'nan,Ma Dongwei,Chen Weiguang,et al.Improving the adsorption behavior and reaction activity of Co-anchored graphene surface toward CO and O2molecules [J].Sensors Actuators B: Chemical,2015,211: 227-234.
[8]Tang Ya'nan,Yang Zongxian,Dai Xianqi.Trapping of metal atoms in the defects on graphene[J].The Journal of Chemical Physics,2011,135(22) : 224704.
[9]Krasheninnikov A,Lehtinen P,Foster A,et al.Embedding transition-metal atoms in graphene: Structure,bonding,and magnetism[J].Physical Review Letters, 2009,102(12) : 126807.
[10]Ambrosi A,Chee S Y,Khezri B,et al.Metallic impurities in graphenes prepared from graphite can dramatically influence their properties[J].Angewandte Chemie International Edition,2012,51(2) : 500-503.
[11]Wang Hongtao,Wang Qingxiao,Cheng Yingchun,et al.Doping monolayer graphene with single atom substitutions [J].Nano Letters,2012,12(1) : 141-144.
[12]Rastegar S F,Peyghan A A,Hadipour N L.Response of Si-and Al-doped graphenes toward HCN: a computational study[J].Applied Surface Science,2013,265: 412-417.
[13]Zhou Miao,Lu Yunhao,Cai Yongqing,et al.Adsorption of gas molecules on transition metal embedded graphene: a search for high-performance graphene-based catalysts and gas sensors[J].Nanotechnology,2011,22(38) : 385502.
[14]Lee Y,Lee S,Hwang Y,et al.Modulating magnetic characteristics of Pt embedded graphene by gas adsorption(N2,O2,NO2,SO2)[J].Applied Surface Science,2014,289: 445-449.
[15]Kresse G,Furthmüller J.Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set[J].Computation Materials Science,1996,6(1) : 15-50.
[16]Kresse G,Furthmüller J.Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set[J].Physical Review B: Condensed Matter and Materials Physics,1996,54(16) : 11169-11186.
[17]Perdew J,Burke K,Ernzerhof M.Generalized gradient approximation made simple[J].Physical Review Letters,1996,77(18) : 3865-3868.
[18]Kresse G,Joubert D.From ultrasoft pseudopotentials to the projector augmented-wave method[J].Physical Review B: Condensed Matter and Materials Physics,1999,59(3) : 1758.
[19]Monkhorst H J,Pack J D.Special points for Brillouinzone integrations[J].Physical Review B: Condensed Matter and Materials Physics,1976,13(12) : 5188-5192.
[20]Henkelman G,Arnaldsson A,Jónsson H.A fast and robust algorithm for Bader decomposition of charge density [J].Computation Materials Science,2006,36(3) : 354-360.
[21]Carlsson J,Scheffler M.Structural,electronic,and chemical properties of nanoporous carbon[J].Physical Review Letters,2006,96(4) : 46806.
[22]Tang Ya'nan,Yang Zongxian,Dai Xianqi.A theoretical simulation on the catalytic oxidation of CO on Pt/graphene [J].Physical Chemistry Chemical Physics,2012,14(48) : 16566-16572.
【中文责编:英子;英文责编:木南】
【材料科学/Materials Science】
Corresponding author: Associate professor Shen Zigang.E-mail: alion100@ sina.com
Citation: Tang Ya'nan,Shen Zigang,Chen Weiguang,et al.Theoretical study on gas sensitivity of modified graphene[J].Journal of Shenzhen University Science and Engineering,2015,32(4) : 365-370.(in Chinese)
Theoretical study on gas sensitivity of modified graphene
Tang Ya'nan,Shen Zigang,Chen Weiguang,Li Chenggang,and Pan Lijun
Quantum Materials Research Center,College of Physics and Electronic Engineering,Zhengzhou Normal University,Zhengzhou 450044,P.R.China
Abstract:We investigate the adsorption of single CO molecule and O2molecule on metal atom decorated graphene(MA-graphene) surfaces by means of the first-principles method based on density functional theory.The results show that vacancy defects in graphene can improve the stability of metal atom absorption,and make the metal atoms be more positively charged due to their losses of electrons,thus facilitating the adsorption of gas molecules.Compared with other MA-graphenes,the single Al and Mo atom embedded graphene sheets exhibit higher sensitivity and selectivity for the O2molecules.Furthermore,the electronic structure and magnetic property of MA-graphene systems can be regulated by the adsorbed gas molecules.Our results validate the reactivity of graphene surface absorbed by single metal atoms and provide a theoretical view for designing new metal-graphene functional devices.
Key words:surface and interface physics; density functional theory; graphene; gas sensitivity; electronic structure; magnetic property; functional device
作者简介:唐亚楠(1981—),男(汉族),河南省鹤壁市人,郑州师范学院讲师、博士.E-mail: yntang2010@ hotmail.com
基金项目:国家自然科学基金资助项目(U1404109) ;河南省教育厅科学技术基金资助项目(14B140019)
doi:10.3724/SP.J.1249.2015.04365
文献标志码:A
中图分类号:O 641
