Marine Metagenome as A Resource for Novel Enzymes
2015-12-21AmniAlmbdiTkshiGojoboriKtsuhikoMinet
Amni D.Alm’bdi,Tkshi Gojobori*,Ktsuhiko Minet
Computational Bioscience Research Center(CBRC),King Abdullah University of Science and Technology(KAUST), Thuwal 23955-6900,Saudi Arabia
REVIEW
Marine Metagenome as A Resource for Novel Enzymes
Amani D.Alma’abadia,Takashi Gojobori*,b,Katsuhiko Minetac
Computational Bioscience Research Center(CBRC),King Abdullah University of Science and Technology(KAUST), Thuwal 23955-6900,Saudi Arabia
Available online 10 November 2015
Handled by Fangqing Zhao
Microbial diversity; Culture-independent studies; Catalysis; Lipase; Biotechnology
More than 99%ofidentified prokaryotes,including many from the marine environment, cannot be cultured in the laboratory.This lack of capability restricts our knowledge of microbial genetics and community ecology.Metagenomics,the culture-independent cloning of environmental DNAs that are isolated directly from an environmental sample,has already provided a wealth of information about the uncultured microbialworld.Ithas also facilitated the discovery ofnovelbiocatalysts by allowing researchers to probe directly into a huge diversity of enzymes within natural microbial communities.Recent advances in these studies have led to a great interest in recruiting microbial enzymes for the development of environmentally-friendly industry.Although the metagenomics approach has many limitations,it is expected to provide not only scientific insights but also economic benefits,especially in industry.This review highlights the importance ofmetagenomics in mining microbial lipases,as an example,by using high-throughput techniques.In addition,we discuss challenges in the metagenomics as an important part of bioinformatics analysis in big data.
Introduction
Recent developments in catalysis have led to a renewed interest in the use of enzymes for the environmentally-friendly industry.Most industrially-relevant enzymes are of microbial origin [1].Identification and isolation of microbial enzymes are thus important steps in improving industrial processes,although only less than 1%of environmental bacteria can be cultivated in the laboratory[2–4].
The current challenging questions have arisen regarding the discovery,identification,and function validation of the uncultured microorganisms.Metagenomics study,which usually starts from the isolation of environmental DNAs without culture,has emerged as an excellent means to study biodiversity and biotechnological applications in certain conditions such as marine environments(Figure 1)[5–7].It provides insights into the genomic pool of microorganisms that are recovered directly from environmental sources.Thus,metagenomics can be used for not only exploring ecological and environmentalpuzzles,but also finding unique biocatalysts with promisingcharacteristics for biotechnological applications[8–10].In particular of the biotechnological applications,metagenome libraries could be screened based on either protein function or nucleotide sequences.
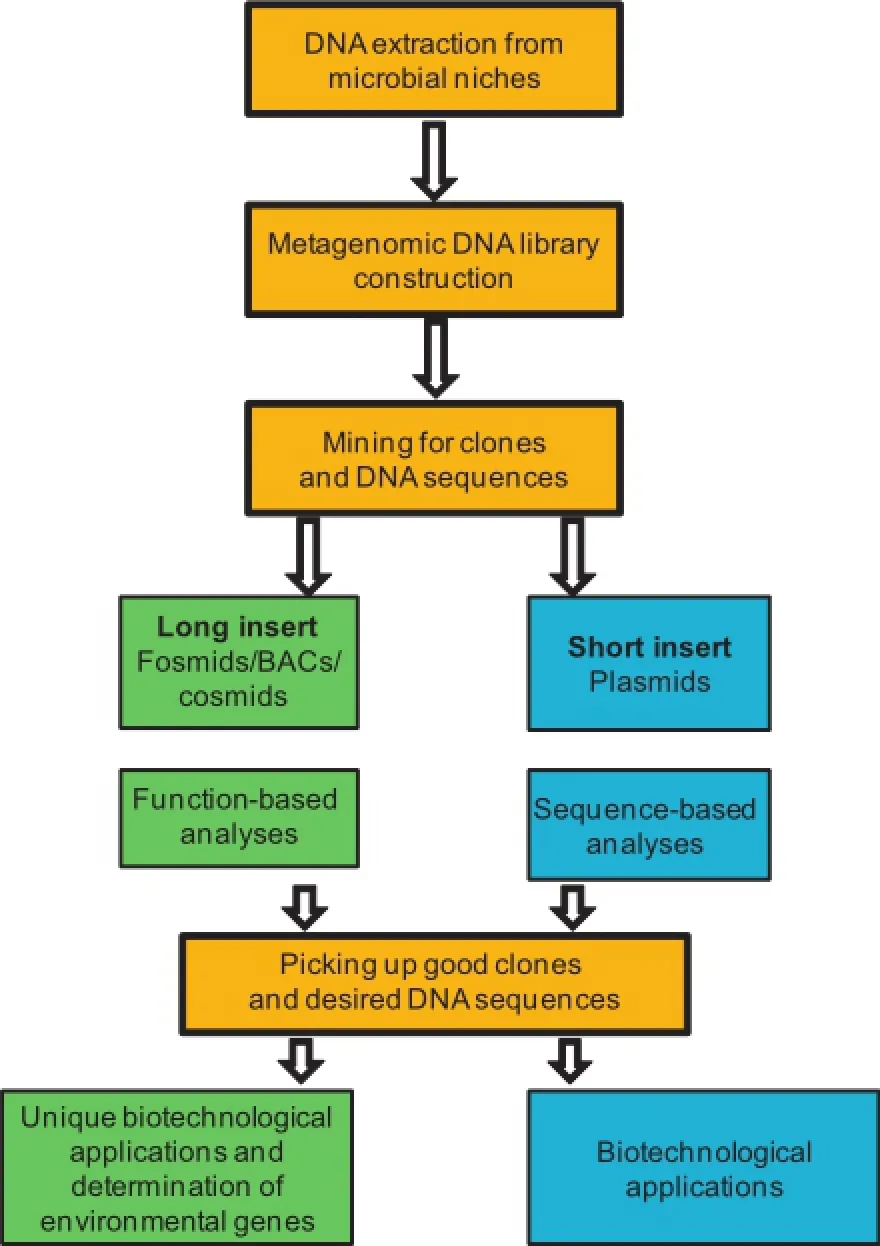
Figure 1 The process of functional metagenomics of marine microbes from environmental samplesThis flowchart illustrates how metagenome is analyzed with the emphasis on the four important processes.BAC,bacterialartificial chromosome.
Function-based screening is a direct way of identifying novel enzymes[2].In this method,enzyme activities are assayed by harvesting a metagenomic library on agar plates enriched with substrates.Positive clones may then be recognized by visual screening for a clear zone called a halo[11]. As a result,function-based screening selects clones with functional activities,such as the synthetic and degradation activities.Unlike sequence-based approaches mentioned later, functional-based screening does not require identification of homologies to genes of known functions.It therefore contributes to nucleic acid and protein databases by adding novel functional annotations.However,this method often suffers from a number of limitations,such as a low hit rate of positive clones,low throughput,and time-consuming screening[11].
In contrast,in sequence-based screening,which involves metagenomic DNA sequencing using next-generation sequencing(NGS)technology,microbial enzymes and bioactive compounds can be explored from niches of interest[10].However, sequence-based screening requires the detection of gene variants with conserved domain or motif of the known functions for enzymes identifications.This approach does not necessarily identify the novel genes.
In light of an increasing demand for enzymes such as carbohydrases,proteases,polymerases,nucleases,and lipases,it is becoming extremely difficult to ignore the importance of hydrolytic enzymes as potential biocatalysts in a wide variety of industries,including chemical processing,dairy,agrochemicals,paper,cosmetics,pharmaceuticals,surfactants,detergents,polymers,and biofuel synthesis[12,13].For example, a lipase is often used at the consumable detergent,as it can hydrolyze fat from clothes and thus enhance its cleaning efficiency.Therefore,the hydrolytic enzymes have been used as promising environmentally-friendly biocatalysts in various industries.
According to the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology,enzymes are classified into six main classes(Table 1).One of the most important classes is hydrolases(E.C.3.-.-.-),which catalyze the hydrolytic cleavage of different types of chemical bonds. Many commercially-critical enzymes belong to this class, e.g.,proteases,amylases,acylases,lipases,and esterases[14]. Lipases are simply hydrolytic enzymes that catalyze hydrolysis and synthesis reactions by breaking down triacylglycerides into free fatty acids and glycerols,which act under aqueous conditions on the carboxylester bonds present in triacylglycerols to liberate fatty acids and glycerol[15–17].Hydrolysis of glycerol esters carrying an acyl chain,which comprises less than 10carbon atoms in length,with tributyrylglycerol(tributyrin)as the standard substrate,usually indicates the presence of an esterase.Most lipases are able to hydrolyze esterase substrates [18].These reactions usually continue with high regio-and/or enantio-selectivity,making lipases a valuable group of biocatalysts in organic chemistry[19–21].

Table 1 Lipase and enzyme classification according to EC number
Two criteria are used to determine if a lipolytic enzyme is a genuine lipase(EC.3.1.1.3)(Table 1).The first criterion is the occurrence of an‘‘interfacial activation”state,which means an increased activity of lipase during emulsion by the triglycerides[18].The second criterion is an active site of the enzymes consisting of a surface loop‘‘lid”that moves together with the interface[17,22,23].These two criteria are important,particularly for the function-based approach.
Various prior studies[24–26]have noted the importance of lipase in the industry,the growing demand for lipases encourages more exploring for novellipases and innovative properties of the known lipases.
Novel lipases found in the environmentalsamples
In recent years,there has been growing interest in finding microbiallipases,principally from bacteria and fungi(Table 2) [27–35].Interestingly,these microorganisms are very attractive as biocatalysts due to their unique properties of adapting to extreme environmental conditions such as hypersaline habitats,high pressure,and extreme temperature.In particular, some microorganisms are able to live in marine environments characterized by high levels of pollutants(e.g.the Norwegian Sea and the Red Sea),high pressures,high temperatures ranging 50–70°C,little to no light or oxygen,and high concentrations of salt and heavy metals[36].As an example of the extreme environmental conditions that are expected to enhance the activities of microbial lipases,we focus on the Red Sea and the microorganisms living there in the present review.
Origin and history of the Red Sea
The Red Sea rift initiated the separation of the African and Arabian(Asian)continent masses about 70 million years ago [37],and the rifting took place multiple times afterward,leading to the eventual formation of the Red Sea.Moreover,frequent episodes of volcanic activities gave rise to the creation of volcanic islands in the Red Sea[37].
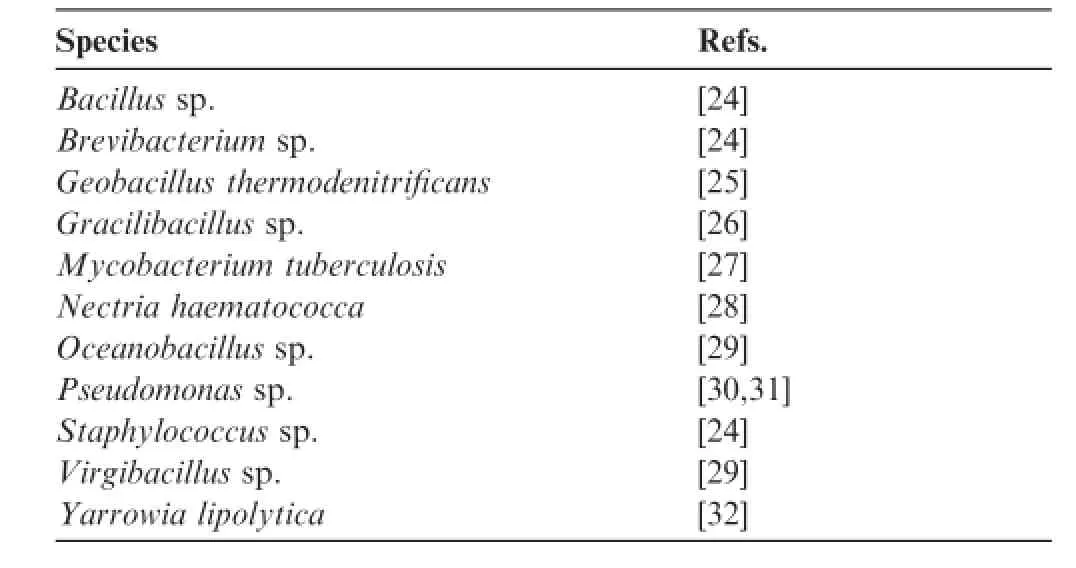
Table 2 List of representative bacterial lipases
The Red Sea’s essential properties make it very unique in the tropics such as no river inflow to the Red Sea.Rainfall is limited between October and May.It is characterized by high salinity,which is estimated in the northern Red Sea to be approximately 40.0 practical salinity unit(psu)[38].The temperature of surface seawater varies from 20°C in spring to 35°C in summer.Since one of the effects by the global warming is the increase in the seawater temperature,thus the Red Sea is attractive to researchers working on climate change. Moreover,a high amount of radiant energy exists throughout the year,reaching its peak in June[39].
The Red Sea has long been considered one of the most diverse and the warmest regions in the world[38,40].It is a geologically young sea basin that has experienced a conversion from a continentalrift to a true oceanic seafloor,producing the high temperature seawater with high concentration of the minerals.Thus,the Red Sea is thought to be an interesting environment to study critical problems such as the microorganism adaptation to the semi-extreme environments as above[41].In addition,these diverse environments with the adaptation can provide a quite different spectrum of microbial diversity in the Red Sea.Thus,the Red Sea can be regarded as a potential source for finding out novel enzymes of lipases.
Challenges in conducting functionalscreening ofmarine metagenomics libraries
Recently,a considerable amount of literature has emerged on isolating lipase enzymes of microbial origin as shown in Table 2.As shown in Figure 1,DNA extracted from environmentalsamples can be cloned into plasmids,fosmids,bacterial artificial chromosomes,or cosmids for proliferation in a suitable heterologous host organism,such as Escherichia coli, and then be screened for catalytic activities.The rapid development of functional screening on metagenomic libraries to find a new enzymatic activity has indicated the importance of microbial diversity in the novel enzyme detection[42–45]. However,the DNA quality of the environmental samples remains a challenge,because of the low copy number of clones and the different insert sizes of metagenomic libraries[46]. DNA purity from marine samples has also been a problem because ofthe complicated and multifarious nature of the marine environments and the role of co-extracted substances,such as humic acids that inhibits biochemical reactions[47].
Currently,choosing the best host system for the construction of heterologous protein and for screening in metagenomic libraries is difficult,because it depends on the nature of target protein,such as a thermo tolerance[48].The gram-negative bacterium,E.coli,is the most used organism for heterologous protein construction as a well-studied model organism.Thus, the E.coli system is the mostly-used host for industrialprotein construction,e.g.,E.coli BL21 and K12[49].However,several types of proteins could not be expressed in E.coli due to the difference of the genetic system in E.coli[50].Thus using alternative bacterial hosts like Bacillus brevis,B.megaterium,and B.subtilis may complement the unachievable goal in E.coli expressing system.B.subtilis and other Bacillus strains were suggested to be the most well-known microbes for the metagenomic libraries screening and heterologous protein construction[50].Gram-positive Bacillus strains have more benefitsin the field of protein production and screening in metagenomic libraries for industrial applications because of absence of lipopolysaccharides in their outer membrane,since lipopolysaccharides in gram-negative bacteria are well-known endotoxin prompting macrophages[51–53].
There are other crucial challenges to be resolved before the full potential of metagenomics can be utilized.First,a huge number of clones are required to be screened as a result of the great biodiversity in the microbial ecosystem.Establishment of high-throughput screening is crucial to identify millions of positive clones in a metagenomic library in a short time.Second,the insert size is a key issue in conducting effective metagenomic screening.For example,a plasmid vector can have a short insert size(less than 10 kb).Therefore,if we use a plasmid as a vector for the library construction,the library harbors only the short length of the target fragments. For this reason,more clones are subsequently required for successfulidentification of positive clones,particularly when compared with metagenomic libraries constructed from fosmid vectors(the insert size is about 40 kb)[7].Furthermore,large clusters of genes cannot be recovered with short inserts.
To overcome the limitations of a cultivation approach,several DNA-based molecular methods have been established, including the capillary-based system of cell culturing on porous hollow-fiber membranes.An analysis of 16S rRNA genes generally supplies considerable information about the species present in an environment[45,54].In particular,various methods can be used to screen novel lipases,including Fourier transform infrared spectroscopy(FTIR).Interestingly,this method has already been used to examine the lipolysis of different substrates(tri octanoyl glycerol and vegetable oils) [55].Hosokawa et al introduced a high-throughput technique for functional screening of a metagenomic library,in which unique enzymes were extracted by droplet-based microfluidics [11].In their method,a microfluidic gel micro-droplet technique was used for co-encapsulation of metagenomic clones to screen a metagenomic library based on a lipolytic activity assay[11].Moreover,they used droplet technology coupled with fluorescent-activated cell sorting to assist the highthroughput screening of enzyme libraries with fluorogenic substrates[56].Thus,powerful techniques such as microfluidics have become a promising tool for screening in metagenomic libraries,especially in selecting novel catalysts.
Metagenomics can be conducted easily to identify genomic segments.However,it is a tough question on how we can obtain a microorganism itself from genomic segments.When a function-based analysis is conducted,it is ideal to have individual samples of the microorganism.This is a serious problem,because most of those microorganisms are uncultivable as mentioned before.To avoid this problem,we may identify a particular coding region in nucleotide sequences that corresponds to a given functional domain of an enzyme such as lipase.Then,using genetic engineering,this functional domain can be expressed to obtain a sufficient amount of proteins in E.coli or yeast for biochemicalassay.If we can invoke a single cell technology;however,isolation of an individual sample of the microorganism of interest is still necessary.This remains one of the biggest challenges.
In short,while metagenomics may help improve our understanding of microbial physiology,genetics,and community ecology[42–44],it can be an advanced and powerful tool for finding out a novel enzyme that is useful for biotechnology application and industrialization.
Challenges related to DNA sequencing and bioinformatics
Over the last few decades,metagenomics has become a fundamental tool in microbial ecology,and a revolution in metagenomic studies is poised to begin,with the support of recent developments in NGS technology.Despite these facts,metagenomics still has computational challenges that need to be addressed.
In the studies of metagenomics,environmental DNA is,in the mostcases,fragmented into smallsegments[57].Therefore, production of millions of small reads must be reassembled de novo utilizing bioinformatics tools and software.However, the reassembly of these reads into contigs is stilla serious computationalchallenge.The reconstruction of the entire genomes of microorganisms in the environmentalsample remains virtually impossible atpresent,although continuous advances in the development of bioinformatics software and tools will have been made.
In fact,depending on the platform used,the read lengths generated from NGS platforms mostly range 75–1000 bp [58].Short read lengths and low depth of coverage lead to the introduction of large gaps in the assembled contigs.Hence, due to the length and number of these gaps,accurate assembly of the contigs is now difficult.Therefore,regeneration of the entire gene sequences becomes extremely difficult and even impossible[59].These challenges can be overcome through a continuous progression of high-throughput gene sequencing technologies and the establishment of methodologies used to sequence longer reads with maximal depth and efficiency. For instance,single molecule real-time sequencing(SMRT) technology,the third-generation sequencing platform,is the latest system developed by Pacific Biosciences[60].This system has the ability to resolve such problems in current gene sequencing platforms by producing longer reads,up to 60 kb with the PacBio RS II platform(http://www.pacificbiosciences.com).Increased depth coverage and long overlapping reads allow reconstruction ofa genome with fewer obstacles,in our prediction.
Instead,there is an approach to annotate the metagenome data without reassembly,i.e.,by classifying the NGS read directly.Functional and taxonomical classifications are the most important processes to revealthe feature of the microbial community and to find the useful enzymes.However,like the assembly of the short reads,the shortness of the NGS reads also prevents the accurate and fast classification.Several software and algorithms to solve this issue such as MetaCV[61] and CVTree3[62]have been developed and main web resources for metagenomics studies are reviewed in this issue as well[63].
Gene prediction is also a challenge in the sequencing of metagenomics data.Many current gene finder systems require long stretches of the sequence to differentiate coding from non-coding sequences.They usually need to train sequences from a single species that is afterward utilized to generate a species-specific gene prediction model[64].Unfortunately,this is inadequate for metagenomes that are constructed from avariety of sequences from distinct microorganisms and frequently constitute not only a limited number of long contigs but also short assemblies and unassembled reads[64]. Moreover,metagenomes are usually permeated with frameshifts that make gene prediction in metagenomes an ambitious task[65].To overcome these issues,the bioinformatics tools to predict genes from metagenome data are actively developed such as MetaGeneMark[66],FragGeneScan[67],and MetaGeneAnnotator[68].
Conclusion
Marine metagenomics is a fast-developing and promising area of genomic studies,by which we can investigate the microbial communities in marine environments.Marine metagenomics has already opened new avenues of research by uncovering a remarkable diversity of marine microorganisms and providing a chance of access to this microbial diversity in laboratory. Marine metagenomics can be used alternatively without culturing microorganisms to discover unique biocatalysts for new functions applied in biotechnological applications,such as lipase enzymes[4,69].In fact,the development of metagenomics has increased the discovery of biocatalysts as many demonstrating novel characteristics.To date,however,most biocatalysts remain uncharacterized.Biocatalysts discovery remains a challenge even with the increased functionalscreening capabilities.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This work was supported by King Abdullah University of Science and Technology(KAUST),Saudi Arabia.
References
[1]Anbu P,Gopinath SC,Chaulagain BP,Tang TH,Citartan M. Microbial enzymes and their applications in industries and medicine 2014.Biomed Res Int 2015;2015:816419.
[2]Uchiyama T,Miyazaki K.Functional metagenomics for enzyme discovery:challenges to efficient screening.Curr Opin Biotechnol 2009;20:616–22.
[3]Amann RI,Ludwig W,Schleifer KH.Phylogenetic identification and in situ detection of individual microbial cells without cultivation.Microbiol Rev 1995;59:143–69.
[4]Jeon JH,Kim JT,Kim YJ,Kim HK,Lee HS,Kang SG,et al. Cloning and characterization of a new cold-active lipase from a deep-sea sediment metagenome.Appl Microbiol Biotechnol 2009;81:865–74.
[5]Simon C,Daniel R.Achievements and new knowledge unraveled by metagenomic approaches.Appl Microbiol Biotechnol 2009;85:265–76.
[6]Simon C,Daniel R.Metagenomic analyses:past and future trends.Appl Environ Microbiol 2011;77:1153–61.
[7]Lopez-Lopez O,Cerdan ME,Gonzalez Siso MI.New extremophilic lipases and esterases from metagenomics.Curr Protein Pept Sci 2014;15:445–55.
[8]Handelsman J,Rondon MR,Brady SF,Clardy J,Goodman RM. Molecular biological access to the chemistry of unknown soil microbes:a new frontier for natural products.Chem Biol 1998;5: R245–9.
[9]Schloss PD,Handelsman J.Biotechnological prospects from metagenomics.Curr Opin Biotechnol 2003;14:303–10.
[10]Steele HL,Streit WR.Metagenomics:advances in ecology and biotechnology.FEMS Microbiol Lett 2005;247:105–11.
[11]Hosokawa M,Hoshino Y,Nishikawa Y,Hirose T,Yoon DH, Mori T,et al.Droplet-based microfluidics for high-throughput screening of a metagenomic library for isolation of microbial enzymes.Biosens Bioelectron 2015;67:379–85.
[12]Hasan F,Shah AA,Hameed A.Industrial applications of microbial lipases.Enzyme Microb Tech 2006;39:235–51.
[13]Saxena RK,Ghosh PK,Gupta R,Davidson WS,Bradoo S, Gulati R.Microbial lipases:potential biocatalysts for the future industry.Curr Sci India 1999;77:101–15.
[14]Zhang C,Kim SK.Research and application of marine microbial enzymes:status and prospects.Mar Drugs 2010;8:1920–34.
[15]Gupta R,Gupta N,Rathi P.Bacterial lipases:an overview of production,purification and biochemicalproperties.Appl Microbiol Biotechnol 2004;64:763–81.
[16]Dalmaso GZ,Ferreira D,Vermelho AB.Marine extremophiles:a source of hydrolases for biotechnologicalapplications.Mar Drugs 2015;13:1925–65.
[17]Su J,Zhang F,Sun W,Karuppiah V,Zhang G,Li Z,et al.A new alkaline lipase obtained from the metagenome of marine sponge Ircinia sp.World J Microbiol Biotechnol 2015;31:1093–102.
[18]Jaeger KE,Dijkstra BW,Reetz MT.Bacterial biocatalysts: molecular biology,three-dimensional structures,and biotechnological applications of lipases.Annu Rev Microbiol 1999;53:315–51.
[19]Mukherjee KD.Plant lipases and their application in lipid biotransformations.Prog Lipid Res 1994;33:165–74.
[20]Jaeger KE,Eggert T,Eipper A,Reetz MT.Directed evolution and the creation of enantioselective biocatalysts.Appl Microbiol Biotechnol 2001;55:519–30.
[21]Jaeger KE,Reetz MT.Microbial lipases form versatile tools for biotechnology.Trends Biotechnol 1998;16:396–403.
[22]Brzozowski AM,Derewenda U,Derewenda ZS,Dodson GG, Lawson DM,Turkenburg JP,et al.A model for interfacial activation in lipases from the structure of a fungallipase–inhibitor complex.Nature 1991;351:491–4.
[23]Derewenda U,Brzozowski AM,Lawson DM,Derewenda ZS. Catalysis at the interface—the anatomy of a conformational change in a triglyceride lipase.Biochemistry 1992;31:1532–41.
[24]Lee LP,Karbul HM,Citartan M,Gopinath SC,Lakshmipriya T, Tang TH.Lipase-secreting Bacillus species in an oil-contaminated habitat:promising strains to alleviate oil pollution.Biomed Res Int 2015;2015:820575.
[25]Rodrigues RC,Fernandez-Lafuente R.Lipase from Rhizomucor miehei as an industrial biocatalyst in chemical process.J Mol Catal B Enzym 2010;64:1–22.
[26]Schmid A,Dordick JS,Hauer B,Kiener A,Wubbolts M,Witholt B.Industrial biocatalysis today and tomorrow.Nature 2001;409:258–68.
[27]Oliveira LC,Ramos PL,Marem A,Kondo MY,Rocha RC, Bertolini T,et al.Halotolerant bacteria in the Sao Paulo Zoo composting process and their hydrolases and bioproducts.Braz J Microbiol 2015;46:347–54.
[28]Balan A,Ibrahim D,Abdul Rahim R,Ahmad Rashid FA. Purification and characterization of a thermostable lipase from Geobacillus thermodenitrificans IBRL-nra.Enzyme Res 2012;2012:987523.
[29]Rohban R,Amoozegar MA,Ventosa A.Screening and isolation of halophilic bacteria producing extracellular hydrolyses from Howz Soltan Lake,Iran.J Ind Microbiol Biotechnol 2009;36:333–40.
[30]Cao J,Dang G,Li H,Li T,Yue Z,Li N,et al.Identification and characterization oflipase activity and immunogenicity of LipL from Mycobacterium tuberculosis.PLoS One 2015;10:e0138151.
[31]Vaquero ME,Prieto A,Barriuso J,Martinez MJ.Expression and properties of three novel fungal lipases/sterol esterases predicted in silico:comparison with other enzymes of the Candida rugosalike family.Appl Microbiol Biotechnol 2015;99:10057–67.
[32]Vakhlu J,Kour A.Yeast lipases:enzyme purification, biochemical properties and gene cloning.Electron J Biotechnol 2006;9:69–85.
[33]Ji X,Li S,Lin L,Zhang Q,Wei Y.Gene cloning,sequence analysis and heterologous expression of a novel cold-active lipase from Pseudomonas sp.PF16.Technol Health Care 2015;23: S109–17.
[34]Ali MS,Ganasen M,Rahman RN,Chor AL,Salleh AB,BasriM. Cold-adapted RTX lipase from antarctic Pseudomonas sp.strain AMS8:isolation,molecular modeling and heterologous expression.Protein J 2013;32:317–25.
[35]Kumar S,Karan R,Kapoor S,S PS,S KK.Screening and isolation of halophilic bacteria producing industrially important enzymes.Braz J Microbiol 2012;43:1595–603.
[36]Barone R,De Santi C,Palma Esposito F,Tedesco P,Galati F, Visone M,et al.Marine metagenomics,a valuable tool for enzymes and bioactive compounds discovery.Front Mar Sci2014. http://dx.doi.org/10.3389/fmars.2014.0003.
[37]Klaus R.Coral reefs and communities of the Central and Southern Red Sea(Sudan,Eritrea,Djibouti,and Yemen).In: Rasul NMA,Stewart ICF,editors.The Red Sea.Berlin Heidelberg:Springer-Verlag;2015.p.409–51.
[38]Berumen ML,Hoey AS,Bass WH,Bouwmeester J,Catania D, Cochran JEM,et al.The status of coral reef ecology research in the Red Sea.Coral Reefs 2013;32:737–48.
[39]Shaikh EA,Roff JC,Dowidar NM.Phytoplankton ecology and production in the Red Sea off Jiddah,Saudi Arabia.Mar Biol 1986;92:405–16.
[40]Stehli FG,Wells JW.Diversity and age patterns in hermatypic corals.Syst Zool 1971;20:115–26.
[41]Rasul NMA,Stewart ICF,editors.The Red Sea.Berlin Heidelberg:Springer-Verlag;2015.
[42]Ferrer M,Martinez-Abarca F,Golyshin PN.Mining genomes and’metagenomes’for novel catalysts.Curr Opin Biotechnol 2005;16:588–93.
[43]Cowan D,Meyer Q,Stafford W,Muyanga S,Cameron R, Wittwer P.Metagenomic gene discovery:past,present and future. Trends Biotechnol 2005;23:321–9.
[44]Cowan DA,Arslanoglu A,Burton SG,Baker GC,Cameron RA, Smith JJ,et al.Metagenomics,gene discovery and the ideal biocatalyst.Biochem Soc Trans 2004;32:298–302.
[45]Streit WR,Schmitz RA.Metagenomics—the key to the uncultured microbes.Curr Opin Microbiol 2004;7:492–8.
[46]Hurt RA,Qiu X,Wu L,Roh Y,Palumbo AV,Tiedje JM,et al. Simultaneous recovery of RNA and DNA from soils and sediments.Appl Environ Microbiol 2001;67:4495–503.
[47]Corinaldesi C,Danovaro R,Dell’Anno A.Simultaneous recovery of extracellular and intracellular DNA suitable for molecular studies from marine sediments.Appl Environ Microbiol 2005;71:46–50.
[48]Angelov A,Mientus M,Liebl S,Liebl W.A two-host fosmid system for functional screening of(meta)genomic libraries from extreme thermophiles.Syst Appl Microbiol 2009;32: 177–85.
[49]Tenaillon O,Skurnik D,Picard B,Denamur E.The population genetics of commensal Escherichia coli.Nat Rev Microbiol 2010;8:207–17.
[50]Westers L,Westers H,Quax WJ.Bacillus subtilis as cell factory for pharmaceutical proteins:a biotechnological approach to optimize the host organism.Biochim Biophys Acta 2004;1694:299–310.
[51]Terpe K.Overview of bacterial expression systems for heterologous protein production:from molecular and biochemicalfundamentals to commercial systems.Appl Microbiol Biotechnol 2006;72:211–22.
[52]Wang H,Bloom O,Zhang M,Vishnubhakat JM,Ombrellino M, Che J,et al.HMG-1 as a late mediator of endotoxin lethality in mice.Science 1999;285:248–51.
[53]Zweers JC,Barak I,Becher D,Driessen AJ,Hecker M,Kontinen VP,et al.Towards the development of Bacillus subtilis as a cell factory for membrane proteins and protein complexes.Microb Cell Fact 2008;7:10.
[54]Stewart EJ.Growing unculturable bacteria.J Bacteriol 2012;194:4151–60.
[55]Beisson F,Tiss A,Riviere C,Verger R.Methods for lipase detection and assay:a critical review.Eur J Lipid Sci Technol 2000;102:133–53.
[56]Aharoni A,Amitai G,Bernath K,Magdassi S,Tawfik DS.Highthroughput screening of enzyme libraries:thiolactonases evolved by fluorescence-activated sorting of single cells in emulsion compartments.Chem Biol 2005;12:1281–9.
[57]Pop M,Salzberg SL.Bioinformatics challenges of new sequencing technology.Trends Genet 2008;24:142–9.
[58]Henson J,Tischler G,Ning Z.Next-generation sequencing and large genome assemblies.Pharmacogenomics 2012;13:901–15.
[59]Riesenfeld CS,Schloss PD,Handelsman J.Metagenomics: genomic analysis of microbial communities.Annu Rev Genet 2004;38:525–52.
[60]Rhoads A,Au KF.PacBio sequencing and its applications. Genomics Proteomics Bioinformatics 2015;13:278–89.
[61]Liu J,Wang H,Yang H,Zhang Y,Wang J,Zhao F,et al. Composition-based classification of short metagenomic sequences elucidates the landscapes of taxonomic and functionalenrichment of microorganisms.Nucleic Acids Res 2013;41:e3.
[62]Zuo G,Hao B.CVTree3 web server for whole-genome-based and alignment-free prokaryotic phylogeny and taxonomy.Genomics Proteomics Bioinformatics 2015;13:321–31.
[63]Dudhagara P,Bhavsar S,Bhagat C,Ghelani A,Bhatt S,Patel R. Web resources for metagenomics studies.Genomics Proteomics Bioinformatics 2015;13:296–303.
[64]Teeling H,Glockner FO.Current opportunities and challenges in microbial metagenome analysis—a bioinformatic perspective. Brief Bioinform 2012;13:728–42.
[65]Hoff KJ.The effect of sequencing errors on metagenomic gene prediction.BMC Genomics 2009;10:520.
[66]Zhu W,Lomsadze A,Borodovsky M.Ab initio gene identification in metagenomic sequences.Nucleic Acids Res 2010;38:e132.
[67]Rho M,Tang H,Ye Y.FragGeneScan:predicting genes in short and error-prone reads.Nucleic Acids Res 2010;38:e191.
[68]Noguchi H,Taniguchi T,Itoh T.MetaGeneAnnotator:detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes.DNA Res 2008;15:387–96.
[69]Turnbaugh PJ,Gordon JI.An invitation to the marriage of metagenomics and metabolomics.Cell 2008;134:708–13.
2 October 2015;revised 19 October 2015;accepted 19 October 2015
.
E-mail:takashi.gojobori@kaust.edu.sa(Gojobori T).aORCID:0000-0001-8895-5423.bORCID:0000-0001-7850-1743.cORCID:0000-0002-4727-045X.
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- CVTree3 Web Server for Whole-genome-based and Alignment-free Prokaryotic Phylogeny and Taxonomy
- Soil and Rhizosphere Associated Fungi in Gray Mangroves(Avicennia marina)from theRed Sea—A Metagenomic Approach
- First Insights into the Viral Communities of the Deep-sea Anoxic Brines of the Red Sea
- Web Resources for Metagenomics Studies
- PacBio Sequencing and Its Applications
- The Value and Significance of Metagenomics of Marine Environments