同位素稀释-气相色谱-三重四极杆串联质谱法分析食用油中18种多环芳烃
2015-12-07陈大舟
许 婷,汤 桦,陈大舟,李 蕾
(1.北京化工大学环境有害化学物质分析北京市重点实验室,北京 100029;2.中国计量科学研究院化学计量与分析科学研究所,北京 100013)
同位素稀释-气相色谱-三重四极杆串联质谱法分析食用油中18种多环芳烃
许 婷1,汤 桦2,陈大舟2,李 蕾1
(1.北京化工大学环境有害化学物质分析北京市重点实验室,北京 100029;2.中国计量科学研究院化学计量与分析科学研究所,北京 100013)
建立了同位素稀释气相色谱-三重四极杆串联质谱测定食用油中18种多环芳烃的方法,同时对前处理过程中固相萃取柱的选择、净化条件及离子源温度等条件进行了优化。样品与环己烷以1∶1(V/V)混匀后,采用分子印迹固相萃取柱串联石墨化碳黑柱浓缩与净化,多反应监测扫描模式下进行质谱检测。结果表明:线性范围为1~200μg/kg时,该方法线性关系良好(R2>0.999),检出限为0.03~0.27μg/kg,定量限为0.10~0.89μg/kg;添加水平在5、10、50μg/kg时,前处理回收率为67.9%~100.8%,精密度(n=3)为0.5%~8.7%。应用该方法检测市场上常见的食用油,未发现食用油中多环芳烃含量超标的现象。该方法可靠性高、速度快、灵敏度高,可用于食用油中多环芳烃的准确检测和标准物质定值。
同位素稀释质谱(ID-MS);多环芳烃(PAHs);食用油;分子印迹固相萃取柱(MISPE);气相色谱-三重四极杆质谱(GC-MS/MS)
多环芳烃(polycyclic aromatic hydrocarbons,PAHs)是一类由2个或2个以上芳香环组成的,具有致畸、致癌、致突变性的一类有机化合物,属于较为常见的食品和环境污染物[1]。在食用油中,多环芳烃从小环到多元环分布较为广泛,长期摄入含有多环芳烃的食用油会危害人们的健康。目前,欧盟规定将食用油中苯并[a]芘与4种多环芳烃的总量(PAH4,苯并[a]蒽、屈、苯并[b]荧蒽、苯并[a]芘的总和)[2]作为食品中多环芳烃含量的指标,其限量分别为2μg/kg和10μg/kg。在我国则将苯并[a]芘作为食用油中多环芳烃含量的指标,其限量为10μg/kg。食用油基体复杂,含有甘油三酯、脂肪酸、游离自醇、石蜡等干扰物,且其中的多环芳烃含量较低,通常为痕量或超痕量级,因此,检测难度大、检测流程复杂,尤其是在样品前处理过程中,降低检查干扰物含量、提高检测灵敏度已成为食用油中多环芳烃检测迫切需要解决的问题。
检测食用油中多环芳烃的前处理技术通常包括液液萃取(LLE)[39]、超声[10]、超临界流体萃取(SFE)[11]、微波辅助萃取(MAE)[12]、固相微萃取(SPME)[13]、凝胶渗透色谱(GPC)[10]、固相萃取(SPE)[3-9,14]等。其中,固相萃取因具有简单、方便、节省时间等优点,被广泛应用于前处理净化中,但常见的固相萃取柱填料对食用油中多环芳烃的选择性较差,净化能力不完全。分子印迹聚合物(molecularly imprinted polymers,MIPs)填料的发展使这一问题得到了改善。分子印迹聚合物对某个或某类化合物具有特异性选择,目前正逐渐应用于固相萃取及固相萃取前处理技术中。分子印迹固相萃取(MISPE)[15]技术可以大幅提高对目标化合物的选择性及分离效率,能同步完成前处理步骤的提取与净化。但是,分子印迹技术的特异性强,对于由2~6个不同个数的芳香环组成的多环芳烃的选择和净化效果差别较大,仍具有一定的局限性。目前,用于多环芳烃分子印迹柱的填料只能对部分四元环及其以上的多环芳烃具有较好的选择性,而对小环多环芳烃的选择性则较差,这是将分子印迹技术应用于食用油中多环芳烃检测的较大障碍。
食用油中多环芳烃的检测方法主要有液相色谱法(LC)[3,11]、气相色谱-质谱法(GC/MS)[4,7,10,14]、气相色谱-三重四极杆串联质谱法(GC-MS/MS)[8]等。相比其他技术,GCMS/MS在多反应监测离子扫描(MRM)模式下,具有更高的灵敏度和选择性,能更大程度地排除基体干扰和基质效应。
本研究采用分析印迹固相萃取柱串联石墨化碳黑柱对样品提取与净化,建立同位素稀释-气相色谱-三重四极杆质谱同时测定食用油中18种多环芳烃的检测方法,旨在为食用油中多环芳烃的准确检测和标准物质定值提供参考。
1 实验部分
1.1 仪器装置与试剂材料
Trace GC气相色谱(配有AS3000自动进样器)、TSQ Quantum XLS三重四极杆质谱仪:美国Thermo-Fisher公司产品;DB-EUPAH气相毛细管色谱柱(20m×0.18mm×0.14μm):
美国J&W公司产品;XS205电子天平(最大量程为81g,分辨率为0.01mg)、UMX5高精密度分析天平(最大量程为5.1g,分辨率为0.1μg):瑞士Mettler Toledo公司产品;Vortex-Genie2T斡旋混合器:美国Scientific Industries公司产品;N-EVAPTMⅢ氮吹仪:美国Oganomation Associates公司产品;Visiprerptm DL固相萃取装置、SupelMIPTMSPE-PAH固相萃取柱(0.5mg/1mL)、SupelCarbTMSPE固相萃取柱(1g/6mL):美国Supelco公司产品。
环己烷、异辛烷、乙酸乙酯(色谱纯):美国Fisher Scientific公司产品;丙酮(色谱纯):美国J.T.Beaker公司产品。16种US EPA PAHs标准混合溶液:由中国计量科学研究院提供;二苯并[a,i]芘、二苯并[a,e]芘(DBaeP)标准品:德国Dr.Ehrenstorfer GmbH公司产品;16种多环芳烃同位素内标:由美国Cambridge Isotope公司提供,其中,D14-二苯并[a,i]芘为D8-甲苯作为溶剂的200mg/L标准溶液,13C6-DBaeP为正壬烷甲苯溶液(80∶20,V/V)作为溶剂的100mg/L标准溶液;D10-蒽:美国ACROS Organics公司产品;D12-苯并[a]蒽:德国Dr.Ehrenstorfer GmbH公司产品。所有标准品的纯度均在98%以上。
食用油:购自北京超级市场;空白油样:为对市场上的食用油进行筛选测定后,选出不含有18种多环芳烃或含量低于2μg/kg的食用油样品。
1.2 实验条件
1.2.1 色谱条件 载气(He,纯度>99.999%)流速:1.0mL/min,保持10min,再以5mL/min升至1.7mL/min;进样口温度300℃,不分流进样;进样量1μL;程序升温:初始温度70℃,保持1min,以30℃/min升至200℃,再以3℃/min升至225℃,然后以4℃/min升至266℃,再以5℃/min升至300℃,最后以10℃/min升至320℃,保持10min。
1.2.2 质谱条件 电子轰击离子源(EI源);离子源温度280℃;电子能量70eV;灯丝发射电流35μA;电子倍增检测器电压1.7kV;碰撞气(氩气)压力0.160Pa;传输线温度300℃;四极杆分辨率:Q1、Q3均为0.7。检测方法:在MRM模式下,针对18种PAHs设定不同的检测窗口,分别测定其离子强度。相应的检测参数列于表1。

表1 18种多环芳烃的保留时间和选择离子反应监测条件Table 1 The retention time and SRM parameters of 18 PAHs
1.3 样品处理
1.3.1 样品制备 在0.5mL食用油中加入0.5mL 10μg/kg的18种多环芳烃内标混合溶液,以环己烷作为溶剂,准确称量,涡旋1min,保证食用油与溶剂及内标充分混合。
1.3.2 固相萃取 用3mL环己烷对串联固相萃取柱活化,串联时,分子印迹柱在上,石墨化碳黑柱在下,柱子保持湿润状态。上样1mL,控制流速,用3mL环己烷淋洗,除去食用油中的杂质,使用空气泵将柱子抽至近干,将串联柱的位置调换,即分子印迹柱在石墨化碳黑柱下面,用干净的安瓿瓶接收洗脱液,用8mL甲苯乙酸乙酯溶液(5∶95,V/V)洗脱目标化合物,将串联固相萃取柱抽干。
1.3.3 氮吹浓缩 在40℃水浴温度下温和氮吹,浓缩至200μL后,转移至2mL小样品瓶中,上机检测。
1.4 实验质量控制
研究过程中发现,小环多环芳烃(2~3个环)在实验器皿中及固相萃取柱上均有残留,由于食用油中多环芳烃的含量较低,因此对广泛存在于实验室中的小环多环芳烃进行干扰排除和空白监测就显得尤为重要。在实验过程中,所用的器皿均水洗烘干后,再用丙酮润洗,实验前再用环己烷润洗。石墨化碳黑柱用15mL甲苯少量多次洗涤,分子印迹柱采用6mL环己烷少量多次洗涤,且所用试剂均为色谱纯。对于玻璃器皿,400℃焙烘10h后备用,使用前用丙酮润洗。测定样品的同时进行全程空白实验,进行比较和扣除。
2 结果与讨论
2.1 前处理条件的优化
2.1.1 固相萃取柱的选择 分子印迹固相萃取柱与传统固相萃取柱相比,具有选择性高、特异性强等特点。因此,采用多环芳烃分子印迹固相萃取柱对食用油中的多环芳烃进行提取与净化,能够特异性识别多环芳烃,有效去除基体中的杂质。将分子印迹柱应用于本研究的18种多环芳烃的净化,能够较好的保留大环多环芳烃,回收率均在73%~95%之间,结果示于图1。但对于小环多环芳烃(2~3个环)和萘、苊烯、苊、芴、菲、蒽及部分4个环的多环芳烃(即荧蒽、芘)的选择性则较差,回收率为19%~51%。
石墨化碳黑固相萃取柱填料的平面片层结构对大环多环芳烃具有永久性吸附作用,但却可以成功地吸附保留上样及淋洗过程中损失的小环多环芳烃,使用适当的溶剂可将一定量的小环多环芳烃洗脱下来。因此,本研究使用石墨化碳黑固相萃取柱串联分子印迹固相萃取柱对食用油中18种多环芳烃进行提取与净化。
2.1.2 固相萃取净化方式的优化 与传统的固相萃取净化方式相同,本研究的串联固相萃取柱提取与净化步骤包括活化固相萃取柱、上样、淋洗杂质、洗脱目标化合物。考虑到石墨化碳黑柱对大环多环芳烃具有永久性吸附的作用,因此串联时分子印迹柱与石墨化碳黑柱的相对位置会对结果产生较大影响。
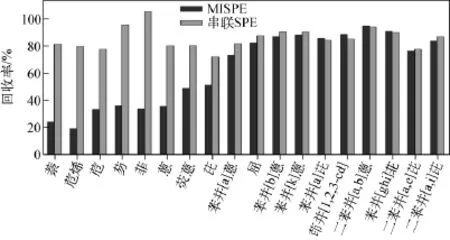
图1 分子印迹固相萃取柱(MISPE)及串联固相萃取柱对18种多环芳烃回收率的比较Fig.1 The recoveries of 18 PAHs on MISPE and coupled columns
采用3mL环己烷对固相萃取柱进行活化,将分子印迹柱置于石墨化碳黑柱上方,上样1mL后,采用3mL环己烷淋洗[16],除去油脂
等杂质。使用空气泵将串联柱抽真空至近干,同时调换固相萃取柱的位置,使分子印迹柱置于石墨化碳黑柱下方,采用8mL甲苯乙酸乙酯溶液(5∶95,V/V)将目标化合物洗脱下来,浓缩后上机检测。结果表明,采用分子印迹柱串联石墨化碳黑柱提取和净化时,18种PAHs回收率在72%~105%之间。小环多环芳烃及个别4元环芳烃的回收率有所提高,且整体回收率均有提高的趋势,这是由于在上样和淋洗时,串联固相萃取柱比单独分子印迹柱的流速慢,使得目标化合物能很好的吸附于固相萃取柱上,达到更好的提取目的。
2.1.3 浓缩过程的优化 前处理中最后一步、也是重要的一个步骤为氮吹浓缩过程。当没有基质化合物吸附多环芳烃时,氮吹至近干的浓缩过程会引起小环多环芳烃78%~98%的损失及部分4元环芳烃(如荧蒽、芘、苯并[a]蒽、屈)20%~48%的损失。为了减少小环多环芳烃的损失,氮吹浓缩过程在40℃水浴中,温和平缓吹至200μL,这样可将多环芳烃在氮吹时的损失控制在4%以内。
2.2 GC-MS/MS条件的优化
2.2.1 扫描时间的优化 扫描时间的缩短可以使色谱峰上扫描点数增加,使峰型更加平滑。但扫描时间过短,会使质量轴发生偏差,导致不能很好的选择定性与定量离子。以苯并[a]芘为例,扫描时间过短时(0.067s),质量轴偏差了m/z0.25。因此,本研究最终将扫描时间设定为0.10s,此时的灵敏度及峰型均能达到较佳状态。
2.2.2 进样口温度和离子源温度的优化 在仪器优化过程中发现,进样口温度较高会降低小环多环芳烃的响应强度,但却可以增加大环多环芳烃的响应强度。因此,本研究对比了进样口温度分别为260、280、300℃时苯并[a]芘的响应强度,每个温度条件下连续进样3针后取平均值,根据所得色谱图的响应强度可知,当进样口温度为300℃时,苯并[a]芘有相对较强的响应值,因此,最终选择300℃作为进样口温度。
除进样口温度外,离子源温度也会对不同多环芳烃的响应强度产生影响。本研究考察了离子源温度为260、280、300℃时苯并[a]芘的响应值,每个温度条件下连续进样3针后取平均值,根据所得色谱图的响应强度可知,随着温度的升高,苯并[a]芘的响应强度增强,当离子源温度升高至300℃时,苯并[a]芘的相对响应值最强。但在实验中发现,离子源的加热棒在300℃时较易损坏,且会加速离子源老化,因此选择280℃作为分析过程中的离子源温度。
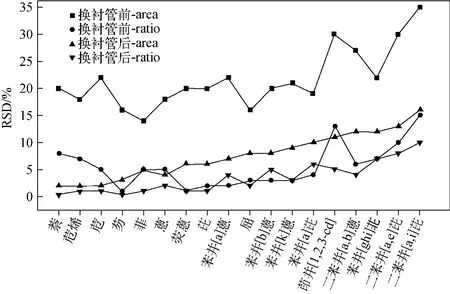
图2 更换衬管前后对18种多环芳烃重复性的影响Fig.2 Effect of lined tube replacement on the RSD of 18 PAHs
2.2.3 衬管对仪器精密度的影响 衬管,尤其是应用于基质样品分析后的衬管,长期使用不仅会降低仪器的灵敏度,还会影响仪器的精密度。实验对比了使用4个月后与新更换的衬管对仪器精密度的影响,结果示于图2。可以看
出:换衬管前,使用各个目标化合物的峰面积来计算的RSD为16%~35%,以目标化合物与相应内标峰面积的比值计算重复性时,RSD为1%~15%;更换新衬管后,精密度明显提高,以峰面积考察时,RSD为2%~15%,比值考察重复性时,RSD为0.3%~9%。同时,在优化前处理过程中发现,以苯并[a]芘为例,衬管使用5个月后,或进样600针以后(包括300针左右的基质溶液和300针的标准溶液),灵敏度降低了68.4%,此时应更换衬管。
2.3 方法验证
2.3.1 标准曲线及检出限 采用称量法分别配制浓度为1、2、5、10、20、50、100、200μg/kg的系列标准溶液,以各物质的质量浓度为横坐标,各物质定量离子响应的峰面积与相应内标响应的峰面积的比值为纵坐标,绘制标准曲线。结果表明,各物质线性关系良好,相关系数R2均大于0.999。以3倍信噪比计算最小检出限(LODs),18种多环芳烃的LODs为0.03~0.27μg/kg;以10倍信噪比计算方法定量限(LOQs),LOQs的范围为0.10~0.89μg/kg,详细结果列于表2。
2.3.2 前处理过程的回收率和精密度 取空白油样样品,分别添加浓度相当于5、10、50μg/kg的18种多环芳烃混合标准溶液,上机前加入混合同位素内标溶液,得到相应的回收率。在该浓度添加水平下,18种多环芳烃的回收率为67.9%~100.8%。按优化的方法分别对3种不同添加浓度的样品进行检测,每种添加浓度的样品重复测定3次,结果列于表3。结果表明,在高、中、低3个浓度添加水平下重复测定3次所得的18种多环芳烃的精密度为0.5%~8.7%。
2.4 实际样品检测
采用本实验的方法测定从市场上采集的4种食用油中的多环芳烃,包括菜籽油、橄榄油、大豆油、花生油,每个样品平行测定3次,结果列于表4。结果表明,所采集的4种食用油中的多环芳烃中,苯并[a]芘的含量均小于2μg/kg,符合我国对食用油中多环芳烃限量的要求;且除二苯并[a,e]芘、二苯并[a,i]芘未检出外,其他16种多环芳烃的方法精密度(n=3)为0.73%~8.11%。
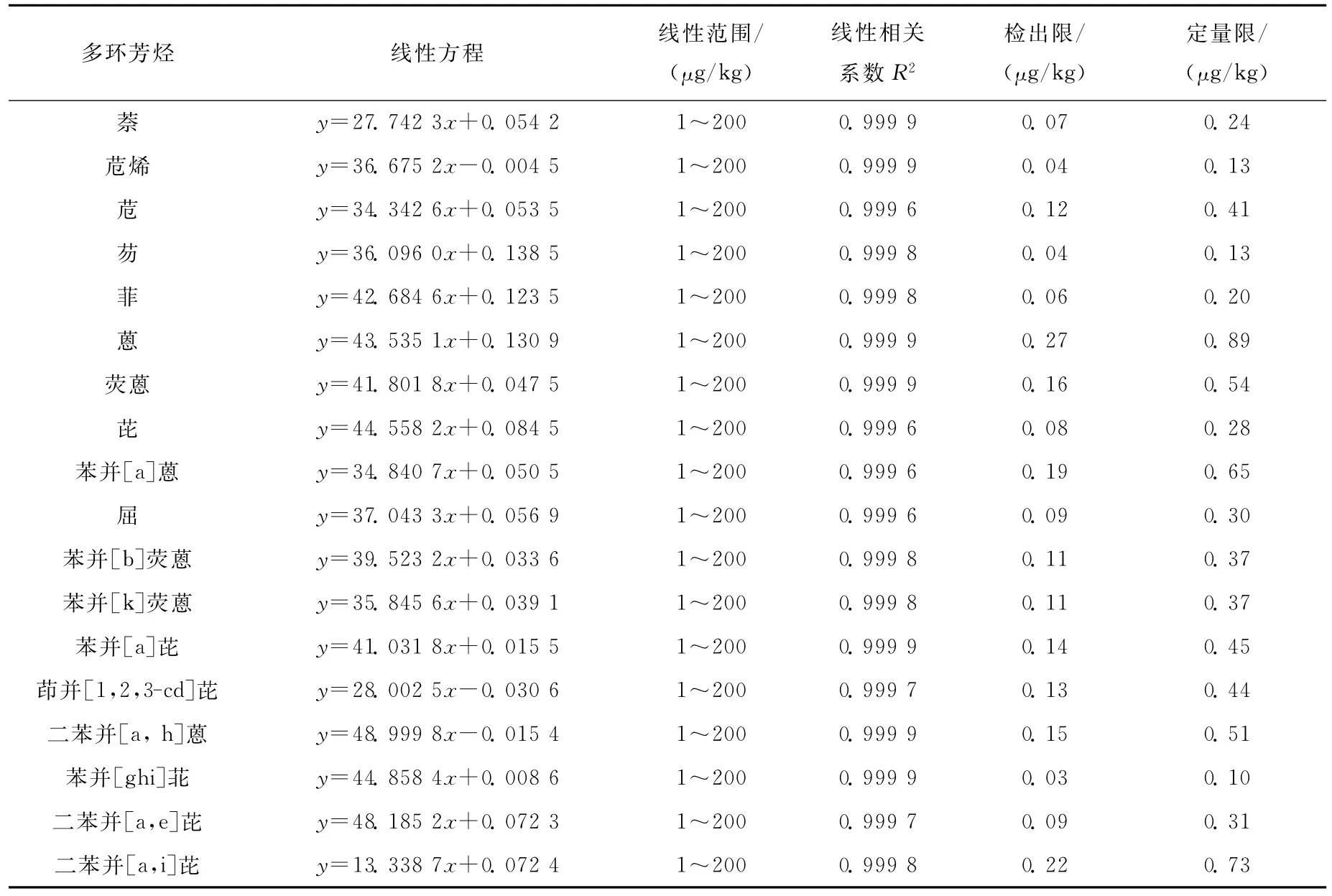
表2 18种多环芳烃的线性方程、检出限与定量限Table 2 Linear equation,LODs and LOQs of the 18 PAHs

表3 18种多环芳烃的回收率及精密度Table 3 Recovery and relative standard deviation(RSD)of spiked edible oil
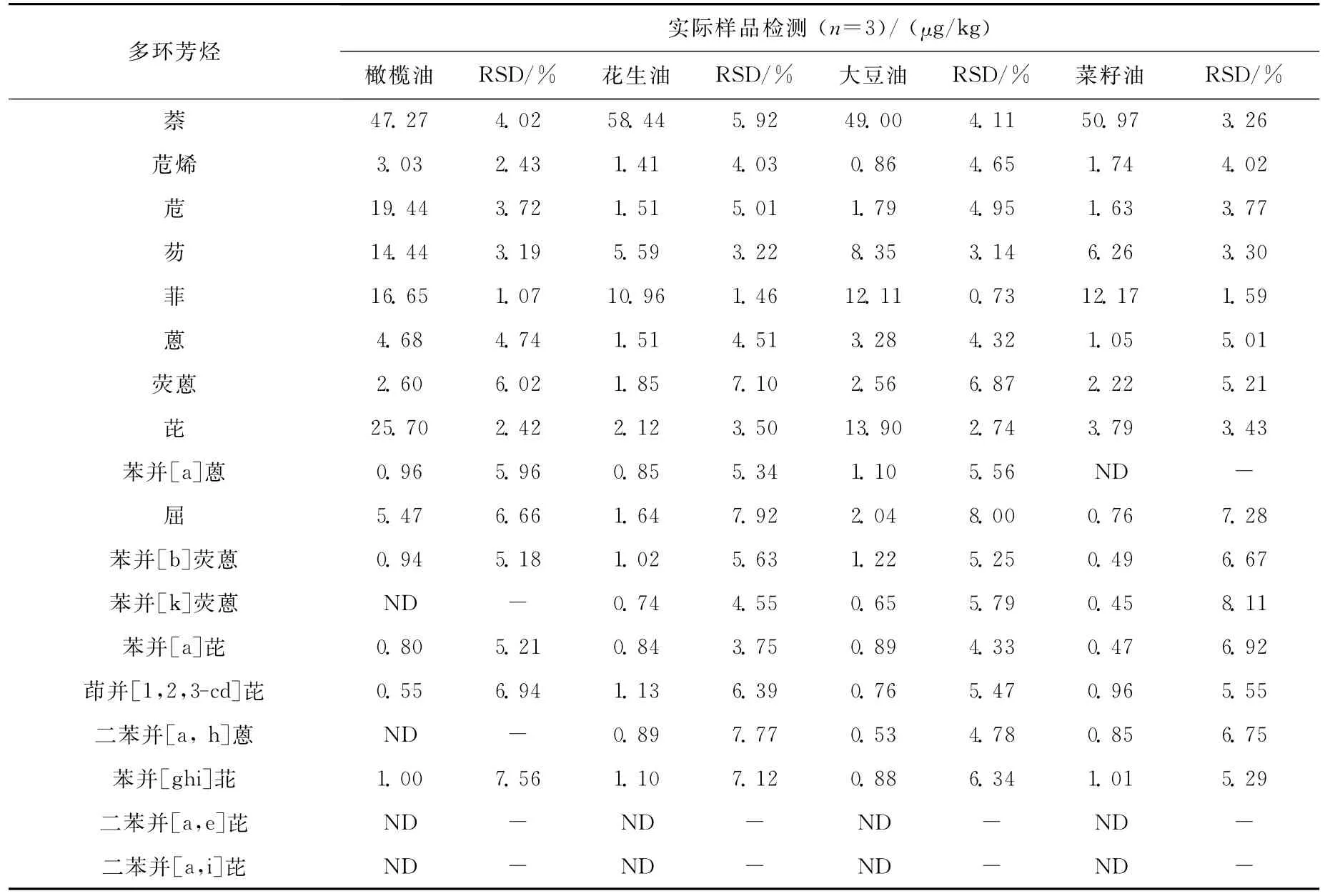
表4 实际样品中多环芳烃的含量检测Table 4 PAHs concentrations in edible oil samples collected at the Beijing market
3 结论
采用同位素稀释气相色谱-三重四极杆质谱法测定食用油中18种PAHs。食用油样品与环己烷混匀后,添加同位素稀释剂,通过分子印迹固相萃取柱串联石墨化碳黑柱提取与净化后,进行浓缩,多反应选择离子扫描方式检测,可以在很大程度上排除基质的干扰,能显著提高测定的选择性和灵敏度。该方法简单、准确、可靠、灵敏度高,可用于食用油样品中痕量PAHs的准确检测和标准物质定值。
[1] FROMBERG A,HØJGÅRD A,DUEDAHLOLESEN L,et al.Analysis of polycyclic aromatic hydrocarbons in vegetable oils combining gel permeation chromatography with solid phase extraction cleanup[J].Food Additives and Contaminants,2007,24(7):758-767.
[2] (EC)835/2011.Maximum levels for polycyclic aromatic hydrocarbons in foodstuffs[S].Official Journal of the European Union,2011.
[3] ROJO CAMARGO M C,ANTONIOLLI P R,et al.Evaluation of polycyclic aromatic hydrocarbons content in different stages of soybean oils processing[J].Food Chemistry,2012,135(3):937-942.
[4] WU S,YU W.Liquid-liquid extraction of polycyclic aromatic hydrocarbons in four different edible oils from China[J].Food Chemistry,2012,134(1):597-601.
[5] CIECIERSKA M,OBIEDZIÑSKI M W.Polycyclic aromatic hydrocarbons in vegetable oils from unconventional sources[J].Food Control,2013,30(2):556-562.
[6] ZHAO W,CHEN X,FANG F,et al.Determination of light-medium-heavy polycyclic aromatic hydrocarbons in vegetable oils by solid-phase extraction and high-performance liquid chromatography with diode array and fluorescence detection[J].Journal of Agricultural and Food Chemistry,2013,61(8):1 804-1 809.
[7] ROSE M,WHITE S,MACARTHUR R,et al.Single-laboratory validation of a GC-MS method for the determination of 27polycyclic aromatic hydrocarbons(PAHs)in oils and fats[J].Food Additives and Contaminants,2007,1 149(6):333-344.
[8] JUNG S Y,PARK J S,CHANG M S,et al.A Simple method for the determination of polycyclic aromatic hydrocarbons(PAH)in edible oil employing solid phase extraction(SPE)cartridge purification[J].Food Science and Biotechnology,2013,22(1):241-248.
[9] BELO R F C,NUNES C M,SANTOS E V D,et al.Single laboratory validation of a SPE method for the determination of PAHs in edible oils by GC-MS[J].Analytical Methods,2012,(4):4 068-4 076.
[10]WANG J H,GUO C.Ultrasonication extraction and gel permeation chromatography clean-up for the determination of polycyclic aromatic hydrocarbons in edible oil by an isotope dilution gas chromatographymass spectrometry[J].Journal of Chromatography A,2010,1 217(28):4 732-4 737.
[11]YUSTY L,CORTIZO DAVIÑA J L.Supercritical fluid extraction and high performance liquid chromatography-fluorescence detection method for polycyclic aromatic hydrocarbons investigation in vegetable oil[J].Food Control,2005,16(1):59-64.
[12]ALARCÓN F,BÁEZ M E,BRAVO M,et al.Feasibility of the determination of polycyclic aromatic hydrocarbons in edible oils via unfolded partial least-squares/residual bilinearization and parallel factor analysis of fluorescence excitation emission matrices[J].Talanta,2013,103:361-370.
[13]PURCARO G,MORRISON P,MORET S,et al.Determination of polycyclic aromatic hydrocarbons in vegetable oils using solid-phase microextraction comprehensive two-dimensional gas chromatography coupled with time-of-flight mass spectrometry[J].Journal of Chromatography A,2007,1 161(1/2):284-291.
[14]AMZAD HOSSAIN M,SALEHUDDIN S M.Polycyclic aromatic hydrocarbons(PAHs)in edible oils by gas chromatography coupled with mass spectroscopy[J].Arabian Journal of Chemistry,2012,5(3):391-396.
[15]ZHENG H B,MO J Z,ZHANG Y,et al.Facile synthesis of magnetic molecularly imprinted polymers and its application in magnetic solid phase extraction for fluoroquinolones in milk samples[J].Journal of Chromatography A,2014,1 329:17-23.
[16]SIGMAEALDRICH.Application Note 192[EB/OL].[2013-09-26].http:∥sigma-aldrich.com/supelco 22.
Determination of 18 Polycyclic Aromatic Hydrocarbons in Edible Oils by Isotope Dilution GC-MS/MS
XU Ting1,TANG Hua2,CHEN Da-zhou2,LI Lei1
(1.Beijing Key Laboratory of Environmentally Harmful Chemical Analysis,Beijing University of Chemical Technology,Beijing100029,China;2.Division of Metrology in Chemistry,National Institute of Metrology P.R.China,Beijing100013,China)
A method for the determination of 18PAHs in edible oils was developed by ID-GC-MS/MS.The oil samples were mixed with cyclohexane in the ratio of 1∶1(V/V)and then cleaned up by molecularly imprinted solid-phase extraction(SPE)catridge coupled to graphitized carbon blacks solid-phase extraction cartridge.Identification and quantification were performed using GC-MS/MS in MRM mode.In the method,the pre-treatment and GC-MS/MS conditions were optimized,such as the selection of SPE columns,clean-up conditions on the tandem columns,the temperature of the ion
isotope dilution mass spectrometry(IDMS);polycyclic aromatic hydrocarbons(PAHs);edible oil;molecularly imprinted polymers solid-phase extraction(MISPE);gas chromatography-tandem mass spectrometry(GC-MS/MS)
O657.63
A
1004-2997(2015)02-0120-08
10.7538/zpxb.youxian.2014.0053
2014-03-11;
2014-04-26
国家质检总局“双打”重点产品检验鉴定技术方法验证评价项目(2012104001)资助
许 婷(1988—),女(汉族),江苏人,硕士研究生,化学专业。E-mail:xuting1477@126.com
李 蕾(1962—),女(汉族),福建人,教授,从事物理化学和环境痕量分析化学研究。E-mail:lilei@mail.buct.edu.cn
时间:2014-08-20;
http:∥www.cnki.net/kcms/doi/10.7538/zpxb.youxian.2014.0053.html
source.The method shows satisfactory linearity(R2>0.999)over the range assayed(1-200μg/kg),and the LODs range from 0.03to 0.27μg/kg,and the LOQs range from 0.10to 0.89μg/kg.The pre-treatment recoveries using this method at three spiked concentration levels(5,10,50μg/kg)range from 67.9%to 100.8%.The RSD is in the range of 0.5%-8.7%.The proposed analytical method has been successfully applied for the analysis of 18PAHs in edible oils obtained from Beijing market.The results indicate that all the detected edible oil samples meet the requirement of China and EU regulation.This indicates that the developed method is suitable for the simultaneous determination and valued of 18PAHs in edible oils.
