Population genetic studies in the genomic sequencing era
2015-12-06HuaCHEN
Hua CHEN
Population genetic studies in the genomic sequencing era
Hua CHEN*
Center for Computational Genomics, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing 100101, China
Recent advances in high-throughput sequencing technologies have revolutionized the field of population genetics. Data now routinely contain genomic level polymorphism information, and the low cost of DNA sequencing enables researchers to investigate tens of thousands of subjects at a time. This provides an unprecedented opportunity to address fundamental evolutionary questions, while posing challenges on traditional population genetic theories and methods. This review provides an overview of the recent methodological developments in the field of population genetics, specifically methods used to infer ancient population history and investigate natural selection using large-sample, large-scale genetic data. Several open questions are also discussed at the end of the review.
Population genetic inference; Allele frequency spectrum; Linkage disequilibrium; Natural selection; Demography
INTRODUCTION
A central goal of evolutionary biology is to understand the mechanisms of how natural selection and other factors, such as random drift and mutation, drive the evolutionary process. Population geneticists address these questions quantitatively by building mathematical models, developing statistical methods for inferring parameters of ancestral processes, and testing hypotheses based on the analysis of real data. With the development of sequencing technologies in recent years, large-sample, large-scale genetic polymorphism data from humans and other species have increased greatly (Mardis, 2008), bringing valuable resources to address evolutionary questions. However, the computational capacities of traditional population genetic methods only allow their applications to small samples and/or local chromosome regions. Methodological development in population genetics has reacted to the emergence of these large-sample, large-scale genetic data. This review provides a summary of methodological advances that aim to answer two fundamental questions in population genetics, specifically ancient demography inference and natural selection detection.
LEARNING ANCIENT DEMOGRAPHY1
In genetic studies of human evolution, mitochondrial DNA (mt-DNA) and the Y chromosome have been invaluable markers for reconstructing the history of modern humans (Cavalli-Sforza & Bodmer, 1971). In their seminal paper, Cann et al (1987) constructed a phylogenetic tree of 147 individuals from five human populations using mt-DNA sequences, and inferred the time of the common female ancestor to be 200 Kyrs. Since mt-DNA is maternally inherited, Cavalry-Sforza and colleagues sequenced the non-recombining region of the Y chromosome (NRY) and developed a system for investigating paternal origins (Underhill et al, 2001). mt-DNA and NRY are especially useful for constructing population history, since the non-recombining regions of mt-DNA or NRY share a single gene genealogy, enabling the inference of a common phylogenetic tree. Using mt-DNA and NRY, population geneticists can determine the migration route and dispersal of global human populations. Many successful applications of this approach can be found in studies of East Asian populations (Jin & Su, 2000; Ke et al, 2001; Kong et al, 2003; Yao et al, 2002; Zhang et al, 2013; Zhao et al, 2009).
Most genetic polymorphisms are, in fact, stored in autosomal regions. Due to recombination along chromosomes, two distant autosomal regions from a sample are likely to evolve with independent genealogies. These gene genealogies represent independent realizations of the evolutionary process given the underlying demographic history, and thus are more informative for demographic inference than mtDNA or NRY, which only carry information of single gene genealogy. One approach to exploring population structure using autosomal polymorphisms is principal component analysis (PCA). This approach decomposes the covariance matrix that summarizes the correlations among individuals or populations, and efficiently extracts the main structure of the data. It then presents the population structure by plotting a few leading principal components against each other. This method was introduced by Cavalli-Sforza & Bodmer (1971) to the field of population genetics, and was very useful for investigating population structure using abundant genomic polymorphism from multiple populations (Patterson et al, 2006).
Recently, other than the exploratory analysis of population structure using PCA, population genetic studies have gained more insights into demographic history from population genetic models. Methods for population history inference have been developed using different aspects of genomic polymorphism, mostly based on: (1) the allele frequency spectrum (AFS, alternatively "site frequency spectrum", SFS), and (2) linkage disequilibrium (LD) or haplotype structure(Table 1).
Allele frequency spectrum-based methods
The AFS is a sampling distribution of alleles in a finite sample (Chen, 2012), and focuses on the allele frequency distribution of a single locus, ignoring the correlation among nearby loci. Such approximation greatly simplifies theory and methodology development. AFS theory was developed in two parallel frameworks: the diffusion (Kimura, 1955) and coalescent processes (Fu, 1995).
Theoretical studies on a single-population AFS started from stationary populations of constant size, and later were extended for populations with time-varying size (Griffiths & Tavaré, 1998), including exponentially growing populations (Evans et al, 2007; Polanski & Kimmel, 2003; Wooding & Rogers, 2002) and piecewise constant populations (see the N-epoch model describing population size change in Marth et al, 2004). The AFS for populations with time-varying size (non-equilibrium populations) was applied to real data to infer ancient Asian and European demography (Keinan et al, 2007).
Recently, the AFS of one population was extended to the joint allele frequency spectrum (JAFS) of multiple populations. Gutenkunst et al (2009) obtained the numerical solution of a three-dimensional diffusion equation, and applied the method to infer the joint demographic history of three world populations: West European (CEU), East Asian (HAN) and West African (YRI). Their softwarehas been extensively applied to analyze genomic data, and uses the finite difference approach to obtain a numerical solution, and thus computational complexity is a function of population size and increases exponentially with population number. The computation becomes intensive when the population number is large or the population is under rapid growth. Lukić et al (2011) reduced computational complexity by using spectral methods, and successfully applied their method to infer the history of four world populations (Lukić & Hey, 2012).
The above two JAFS approaches rely on numerical solutions of diffusion equations. Others have used coalescent simulations to approximate the JAFS of two or several populations (Excoffier et al, 2013; Li & Stephan, 2006). Chen (2012) derived an analytical form of the JAFS for multiple populations using coalescent theory. Their method incorporated various scenarios including time-varying population size, instantaneous migration and the hitch-hiking effect. Compared to the diffusion-based approach, computational complexity of the coalescent-based method is reduced to a function of sample size, which is much more efficient.
Haplotype structure-based methods
Another group of methods consider linkage disequilibrium, or the correlation of gene genealogies of adjacent sites. For example, the pairwise sequential Markovian coalescent method (PSMC, Li & Durbin, 2011) uses a hidden Markov model to approximate the dependency of the coalescent times of two haplotypes between adjacent loci, and further infers the detailed ancient population size from coalescent times. The limitation of the PSMC method is that it can only analyze one diploid genome. Burgess & Yang (2008) inferred ancient population size by analyzing multiple sequences, with each sequence representing one population. They developed a Markov chain Monte Carlo approach (MCMCCoal) to sample gene genealogies. Gronau et al (2011) modified MCMCCoal to allow two sequences from each sampled population, and applied their G-PhoCS method to infer the joint demography of four world populations. The Coal-HMM method (Hobolth et al, 2007) can also analyze multiple genomes from several populations. Instead of sampling over gene genealogies as per MCMCCoal, Coal-HMM treats the unobserved gene genealogy at each genomic position as unobservable latent states in a hidden Markov model. The method in Mailund et al (2011) is similar to the PSMC method, but can analyze two sequences from two populations.
Extending the above methods to the analysis of a large or even medium number of individual genomes is more challenging. Lohse et al (2011) used a probability generation function to infer the coalescent times of multiple individuals, and then population demography. Harris & Nielsen (2013) investigated the extent of shared IBD (identical by descent) tracts between each pair of haplotypes, and applied their method to a two-isolated-population model with migration. Themethod in Sheehan et al (2013) was built on the sequential Markovian coalescent process and improved computation by proposing more efficient importance-sampling proposal distributions. Recently, Schiffels & Durbin (2014) extended Li and Durbin's PSMC method to the multiple sequential Markovian coalescent (MSMC) method, which can deal with multiple individual genomes from two populations. The possible number of gene genealogies increases dramatically when multiple sequences are included, and computation again becomes intensive. MSMC tackles the problem by focusing only on some summary statistics of the genealogies, such as first coalescent time of any two sequences and total length of all singleton branches of the genealogy.
Overall, the existing haplotype-based methods can analyze a small number of individual genomes, but are quite powerful for inferring ancient history. For example, the PSMC method works well for learning population size between 20–200 Kyr (Li & Durbin, 2011). The remaining challenge is to efficiently approximate the sequential Markovian coalescent or the ancestral recombination graph for larger samples.
Demography of East Asian populations
The AFS and haplotype-based methods have been applied to genomic data to infer demographic histories of humans and other species. In human studies, the demographic history of Western Europeans was the main focus due to the abundance of sequence data for European populations. Several studies inferred East Asian demographic history using the HapMap or the Thousand Genomes Project data (Gravel et al, 2011; Gronau et al, 2011; Li & Durbin, 2011; Schaffner et al, 2005; Schiffels & Durbin, 2014). The inferred parameters of these studies are detailed in Table 2. These studies identified major events in East Asian history; for example, Keinan et al (2007) analyzed the AFS of the HapMap HCB samples and estimated a severe bottleneck in East Asian populations with an intensity (defined as T/2N) of 0.123±0.015. While detailed knowledge on Asian population history is still limited, understanding East Asian demographics at a finer level is useful for disease studies, and relies on the availability of genomic polymorphism data.
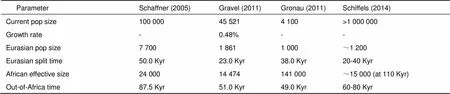
Table 2 Inferred demographic history of East Asians by different studies
DETECTING NATURAL SELECTION
Methods
Detecting the effects of natural selection and identifying the selected loci in the genome is another research hotspot in population genetics. Natural selection, especially positive selection (aka, selective sweeps) generates distinctive genetic polymorphism patterns in contemporary populations. Statistical approaches have been constructed using some informative aspects of these patterns.
Allele frequency spectrum When natural selection drives a selected mutant to fixation in a population, the allele frequencies of SNPs in the vicinity of the selected mutant are also affected. Their frequencies can be increased, if the alleles are linked to the selected mutant, or decreased otherwise. This is known as the hitch-hiking effect. Smith & Haigh (1974) provided a deterministic equation to approximate this effect (Fay & Wu, 2000). Recent theoretical studies on the selective coalescent process derived more accurate sampling formulas for modeling the hitch-hiking effect (Durrett & Schweinsberg, 2004; Etheridge et al, 2006). The sampling formulas can be used to derive the AFS of a linked SNP under hitch-hiking. Methods for detecting selection can then be constructed by combining information of multiple SNP loci with a composite-likelihood approach. As an approximation of the full likelihood, the composite likelihood of multiple loci is obtained by taking the product of marginal likelihood of individual loci. The AFS-based methods include SweepFinder for a single population (Nielsen et al, 2005), and the JAFS method of multiple populations (Chen, 2012).
Summary statistics, such as, Tajima's(Tajima, 1989), Fu and Li's* (Fu & Li, 1993) and Fay and Wu's(Fay & Wu, 2000) are known as neutral tests for selection. These statistics are summaries derived from a single-population AFS. For example, a negativevalue indicates a skewed AFS with overrepresented singletons, while a positiveindicates an enrichment of segregating sites with medium frequencies. A likelihood approach using a detailed AFS has more power than neutral tests based on summary statistics.
AFS methods are powerful for detecting selection, but have several limitations: (1) population history can confound the effect of selection on the AFS. For example, a recent rapid population growth increases the relative abundance of rare alleles, which is similar to the effect of negative selection. However, since demographic effects are genome-wide, it is possible to control for these effects by explicitly modeling demographic history in the methods (Li & Stephan, 2006; Williamson et al, 2005); (2) AFS methods are not robust to SNP ascertainment bias (Clark et al, 2005). SNP data generated from platforms designed under complex ascertainment schemes, such as SNP arrays, are unfit for AFS methods. With more ascertainment-free genomic data from NGS technology, AFS-based methods are expected to become more applicable.
Haplotype structure When a selected allele is increased to high frequency, the ancestral haplotypes carrying the selected alleles are also increased. The time interval for the selection process is short enough that the ancestral haplotypes are not broken by recombinations, and thus long extent haplotypes in the vicinity of the selected allele can be observed. Such a haplotype structure can be used to test for recent positive selection when the selected allele is still segregating. Several methods were developed based on such haplotype structure, including the EHH test (Sabeti et al, 2002), iHS test (Voight et al, 2006), and the hidden Markov model approach (Chen et al, 2015). The idea was further extended to the comparison of haplotype homozygosity between two populations (e.g., XPEHH test, Sabeti et al, 2007; Tang et al, 2007). These haplotype-based methods are robust to SNP ascertainment schemes, and are very useful for SNP array data designed under complicated ascertainment schemes.
Population differentiation Using population differentiation to detect selection is based on the fact that if the gene is under local adaptation in one population, its frequency divergence among populations should be highly beyond the genomic level. The fixation index,st, which measures the divergence between two populations at a single locus, was adopted by Lewontin & Krakauer (1973) to detect selection. A moments-based estimator ofst developed by Weir & Cockerham (1984) has been commonly used when sample sizes from two populations are unbalanced and has been applied to a genome scan for selected loci (Akey et al, 2002).
st is calculated for a single locus and has a large variance. The random fluctuation ofst values across SNP loci causes high false positive rates. Combining the incremental effects of selection on multiple loci helps reduce the false positive rates and increase power. The XP-CLR test by Chen et al (2010) was developed in light of this principle. It explicitly models the decay of population differentiation as a function of genetic distance between the neutral marker and selected mutant, and uses a composite likelihood scheme to combine the effects of multiple loci. Other methods that make use of population differentiation include the locus-specific branch test (LSBL) and its variants (Shriver et al, 2004; Yi et al, 2010), though they are single locus-based.
The above three classes of methods utilize different aspects of data patterns. Grossman et al (2010) attempted to combine multiple genetic signals to reduce the false positive rates in detecting selection (composite of multiple signals test, CMS).
Natural selection in East Asians
Modern humans faced environmental changes and infectious diseases when they migrated out of Africa and colonized other places in the world. Natural selection was very likely essential during this process. Specifically, East Asian environments were extremely divergent in terms of climatic factors, such as UV light, temperature and altitude, making Asian populations ideal for studying natural selection (Shi & Su, 2011).
Shi et al (2009) hypothesized that thepathway may be important for the adaptation to low temperature when East Asians moved from the south towards high-latitude areas. Tibetans have lived on the Himalayan Plateau for tens of thousands of years. Recent genomic studies identified several genes conferring hypoxia adaptation, among whichshows the strongest signal of Tibetan-specific selection (Beall et al, 2010; Peng et al, 2011; Simonson et al, 2010; Wang et al, 2011; Xu et al, 2011a; Yi et al, 2010). Xiang et al (2013) recently identified one functional mutant in another gene. Interestingly,andare both of the hypoxia pathway with direct interaction.
shows an extremely strong signal of recent positive selection in East Asian (Sabeti et al, 2007). Kamberov et al (2013) used mice to show that a non-synonymous mutation could cause phenotypical changes in the skin. The mechanism underlying selection onremains unclear, though it was hypothesized to be due to the high humidity in Eastern Asia.
Life styles, such as the transition from hunter-gather to an agricultural society might also be important driving forces. One gene related to lifestyle transition is(Li et al, 2008; Peng et al, 2010). The*allele in East Asians shows signals of selection and its geographic frequency distribution is consistent with cultural relic sites of rice domestication in China, indicating that thegene might be related to the potential benefits of fermented beverages and food.
Artificial selection in domestication
Domestication and animal and plant breeding have been ongoing since the origin of agriculture around 10000 years ago. Artificial selection shaped the traits of domesticated species to meet human demands during this process (Li & Zhang, 2009). Investigating selection on domestication traits helps identify the genetic architecture of these traits and improve breeding (Doebley et al, 2006). In recent years, such endeavors have been facilitated by genomic sequencing technology (Doebley et al, 2006; Fang et al, 2009; Huang et al, 2012; Hufford et al, 2012; Lyu et al, 2013, 2014; Qi et al, 2013a; Xu et al, 2011b; Zhou et al, 2015).
Most such studies identified a list of selected loci, and then checked their overlap with domestication QTL loci or some meaningful GO categories and pathways. Xia et al (2009) sequenced 40 domesticated and wild silkworms. They identified signals of selection at 354 candidate genes possibly important during domestication, some of which have enriched expression in the silk gland, midgut and testis. Wang et al (2013) studied natural selection in dog genomes, and identified genes related to starch digestion and metabolism, including nutrient transport and regulation of the digestion process. This reflects an agricultural living condition during the domestication history of dogs.
Zhou et al (2015) analyzed genomic sequences of 62 wild soybeans, 130 landraces and 110 improved cultivars. They identified 121 domestication-selective sweeps and 109 improvement-selective sweeps. Among these selected targets, some were related to morphological features, such as seed size and color, seed weight, stem determinacy, flower color, seed coat color and pubescence form. In addition to morphological traits, more than 90 sweep targets were located within known oil QTL regions.
Results from these genomic studies shed light on the fundamental mechanisms of the artificial selection process. For example, Hufford et al (2012) analyzed genomic sequences of 75 wild landraces and improved maize, and found evidence for stronger selection during domestication than improvement and that artificial selection was common in regulatory regions, which was confirmed by transcriptome analysis.
Inferring selection intensity, allele age and fixation time
In addition to identifying loci under selection, population geneticists are also interested in knowing further details of the selection process, such as, when natural selection initialized and how to quantify selection intensity. A detailed portrait of the selection process provides hints for deciphering the mechanism of selection, and validates anthropological hypotheses. For example, in studies on Tibetan high altitude adaptation, Peng et al (2011) and Xiang et al (2013) inferred the allele ages ofandusing haplotype structure. Although both genes were under strong selection, the estimated times were different: selection onstarted around 20 Kyr ago and selection time ofwas only about 7 Kyr. Interestingly, the two selection times are consistent with the two waves of population migrations to the Tibetan Plateau (Qi et al, 2013b; Zhao et al, 2009). Based on this, Xiang et al (2013) proposed a two-step hypothesis on the development of Tibetan adaptation to high altitude.
For selected alleles still segregating in the population, e.g.,and, Chen & Slatkin (2013) proposed a method for inferring selection intensity and allele age using haplotype structure. Their method relies on importance sampling algorithms to sample from the genealogical space and allele frequency trajectories, which requires very intensive computation. The method can only analyze a local region for a small number of individuals. Chen et al (2015) developed a hidden Markov model for investigating the haplotype structure around the selected mutant, and provided a simplified population genetic model for inferring the parameters. The simplified model is much more efficient, and can be applied to genome-wide analysis for large samples.
If selection occurred anciently, the selected allele may have been fixed in the population. The parameter of interest for ancient selection is the time since fixation. For example, the genetic changes underlying the emergence of speech and language in modern humans, believed to be under strong selection, were inferred to be fixed during the last 200 Kyrs (Enard et al, 2002). Linnen et al (2009) studied cryptically colored deer mice living on the Nebraska Sand Hills and showed that their light coloration was caused by a cis-acting mutation closely linked to a single amino acid deletion in. The fixation time of the mutant was 8-10 Kyrs ago.
To date, only a few methods for inferring fixation time of selection have been proposed. The Bayesian approach by Przeworski (2003) simulates samples under selection, and matches a list of summary statistics between simulated and real data with rejection sampling. The method integrates over all possible values of selection intensity, and provides the posterior distribution of fixation time. Linnen et al (2009) used a two-step scheme to infer fixation time, first assuming the fixation time was 0 and estimating the selection intensity using the AFS of all SNPs from the gene region (Kim & Stephan, 2002). Fixing selection intensity to the estimated value, they used the Bayesian approach of Przeworski (2003) to obtain the posterior distribution of fixation time. However, the above two methods fail to jointly estimate selection intensity and fixation time. Chen (2012) modeled the pattern of the allele frequency spectrum of SNPs linked to a selected mutant as a function of selection intensity and fixation time, and efficiently estimated both parameters.
CHALLENGES AND FUTURE STUDIES
With the rapid development of sequencing technology, large-sample or even population-scale sequencing data have become available. For example, Coventry et al (2010) sequenced two genes (and) for 10422 European Americans and 3293 African Americans. Nelson et al (2012) and Fu et al (2013) conducted exome sequencing for several thousand individuals. Large sample genomic data provide valuable resources for population genomic studies. However, the computational capacities of traditional population genetic methods only allow their application to local regions and/or small samples. Developing computationally efficient methods capable of analyzing large-sample, large-scale genomic data is necessary and challenging. Some computational issues, such as controlling for data quality, especially for sequencing data with low coverage, are important for population genetic inference, but beyond the scope of this review. Discussions on this topic can be found in the literature (e.g., Han et al, 2014; Jiang et al, 2009; Johnson & Slatkin, 2006; Liu et al, 2010).
Computational challenges
One main obstacle prohibiting existing population genetic methods from application to large genomic data is intensive computation. The likelihood function of most population genetic methods has gene genealogy as a nuisance variable. These methods evaluate the likelihood function by adopting Markov chain Monte Carlo (MCMC) or importance sampling (IS) to integrate over the gene genealogy space. It is computationally very intensive, and only works for a small sample of haplotypes from a local chromosome region (Griffiths & Tavare, 1994). Such methods cannot be directly scaled up to large-sample genomic data even with high performance computers. Developing efficient computing algorithms is necessary.
Another issue is numerical instability. For example, an essential component in coalescent-based methods is the distributions of coalescent time and ancestral lineage numbers. Both equations are expressed as a function of alternating hypo-geometric series. When the sample size is large (e.g.,>100), the coefficient of each individual term of the series becomes so large that it is beyond the capacity of double-precision variables of any computer language. One scheme to avoid such numerical overflow is to use a high-precision arithmetic library (HPAL) in programming. This significantly increases programming difficulty and computing time with only a limited improvement in performance. A more applicable solution is to replace the exact distributions with their asymptotic distributions (Chen & Chen, 2013; Griffiths, 1984). Asymptotic formulas are usually in simple analytical form and easy to calculate.
Methods for detecting soft sweeps and polygenetic selection
Some questions are raised from a biological view instead of computational issue. One such issue is the revision of our views on the general forms of natural selection. After more than ten years of genome-wide studies on selective sweeps in humans, only a few genes have been identified under strong selection due to extreme environmental factors () or infectious diseases (). This is in conflict with our traditional understanding and urges us to reflect and explore the actual general form of adaptation in nature. Recently, population geneticists hypothesized that other forms of selection, such as, soft sweeps and polygenetic selection, are likely to be more common in nature and are under the radar of existing genomic approaches (Pritchard et al, 2010; Wollstein & Stephan, 2015).
Most conventional methods for detecting selective sweeps were developed by assuming selection starts from amutation. Such a selective process is called a hard sweep. If selection starts from a standing mutant, which has been in the population under neutrality for a long time and is in high frequency, it is a soft sweep (Hermisson & Pennings, 2005). Researchers hypothesized that soft sweeps are more prevalent than hard sweeps (Pritchard & Di Rienzo, 2010). The chance for a new advantageous mutant to occur is very small, and it is also very unlikely that the new advantageous mutant can survive the effect of random drift in the early stage of the selective process to finally reach high frequency in the population. Ohta & Kimura (1975) already noticed this in their seminal paper on the hitch-hiking effect: “It is likely that the new advantageous allele will be chosen, in response to environmental changes, from the pre-existing alleles rather than occurring by mutation”.
Although soft sweeps are more common, it is not trivial to propose a powerful method for detecting soft sweeps. The genetic polymorphism pattern caused by soft sweeps is indistinguishable from that under neutrality in many aspects, including the allele frequency spectrum, reduction of genetic diversity, and linkage disequilibrium. This explains very few methods for detecting soft sweep so far (Przeworski et al, 2005; but see Garud et al, 2015).
To date, genomic studies on selection have focused on a single locus, for example, lactase persistence. Some traits, such as skin pigmentation, are determined by several major genes and show an evolutionary mode similar to the single-gene cases. However, most traits are quantitative and determined by multiple genes with minor effects and complex interactions. “It seems likely to us that, as in traditional quantitative genetic models, many -- possibly even most -- adaptive events in natural populations occur by polygenic adaptation” (Pritchard & Di Rienzo, 2010). Such traits, when under natural selection, tend to evolve in a polygenic mode: one could expect that multiple functional loci shift their allele frequencies without being fixed when the population fitness is improved by natural selection (Hancock et al, 2010). Our understanding of polygenic selection is in the early stages, and as pointed out by Pritchard & Di Rienzo (2010), empirical study and theoretical modeling are both needed to understand the mechanism of polygenic selection.
ACKNOWLEDGEMENTS
I am grateful to Professor Bing SU for hosting the visit to the State Key Lab of Genetic Resources and Evolution and Kunming Institute of Zoology. The author was supported by the CAS 100 Talents Program.
REFERENCES
Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. 2002. Interrogating a High-density SNP map for Signatures of Natural Selection.12(12): 1805-1814.
Beall CM, Cavalleri GL, Deng LB, Elston RC, Gao Y, Knight J, Li CH, Li JC, Liang Y, McCormack M, Montgomery HE, Pan H, Robbins PA, Shianna KV, Tam SC, Tsering N, Veeramah KR, Wang W, Wangdui P, Weale ME, Xu YM, Xu Z, Yang L, Zaman MJ, Zeng CQ, Zhang L, Zhang XL, Zhaxi P, Zheng YT. 2010. Natural selection on() associated with low hemoglobin concentration in Tibetan highlanders.107(25): 11459-11464.
Burgess R, Yang ZH. 2008. Estimation of hominoid ancestral population sizes under Bayesian coalescent models incorporating mutation rate variation and sequencing errors.25(9): 1979-1994.
Cann RL, Stoneking M, Wilson AC. 1987. Mitochondrial DNA and human evolution.325(6099): 31-36.
Cavalli-Sforza LL, Bodmer WF. 1971. The Genetics of Human Populations. San Francisco: Dover Publications.
Chen H. 2012. The joint allele frequency spectrum of multiple populations: A coalescent theory approach.81(2): 179-195.
Chen H, Chen K. 2013. Asymptotic distributions of coalescence times and ancestral lineage numbers for populations with temporally varying size.194(3): 721-736.
Chen H, Patterson N, Reich D. 2010. Population differentiation as a test for selective sweeps.20(3): 393-402.
Chen H, Hey J, Slatkin M. 2015. A hidden Markov model for investigating recent positive selection through haplotype structure.99: 18-30.
Chen H, Slatkin M. 2013. Inferring selection intensity and allele age from multi-locus haplotype structure.3(8): 1429-1442.
Clark AG, Hubisz MJ, Bustamante CD, Williamson SH, Nielsen R. 2005. Ascertainment bias in studies of human genome-wide polymorphism.15(11): 1496-1502.
Coventry A, Bull-Otterson LM, Liu XM, Clark AG, Maxwell TJ, Crosby J, Hixson JE, Rea TJ, Muzny DM, Lewis LR, Wheeler DA, Sabo A, Lusk C, Weiss KG, Akbar H, Cree A, Hawes AC, Newsham I, Varghese RT, Villasana D, Gross S, Joshi V, Santibanez J, Morgan M, Chang K, Hale Iv W, Templeton AR, Boerwinkle E, Gibbs R, Sing CF. 2010. Deep resequencing reveals excess rare recent variants consistent with explosive population growth.1(8): 131.
Doebley JF, Gaut BS, Smith BD. 2006. The molecular genetics of crop domestication.127(7): 1309-1321.
Durrett R, Schweinsberg J. 2004. Approximating Selective Sweeps.66(2): 129-138.
Enard W, Przeworski M, Fisher SE, Lai CSL, Wiebe V, Kitano T, Monaco AP, Pääbo S. 2002. Molecular evolution of, a gene involved in speech and language.418(6900): 869-872.
Etheridge A, Pfaffelhuber P, Wakolbinger A. 2006. An approximate sampling formula under genetic hitchhiking.16(2): 685-729.
Evans SN, Shvets Y, Slatkin M. 2007. Non-equilibrium theory of the allele frequency spectrum.71(1): 109-119.
Excoffier L, Dupanloup I, Huerta-Sánchez E, Sousa VC, Foll M. 2013. Robust demographic inference from genomic and SNP data.9(10): e1003905.
Fang MY, Larson G, Ribeiro HS, Li N, Andersson L. 2009. Contrasting mode of evolution at a coat color locus in wild and domestic pigs.5(1): e1000341.
Fay JC, Wu CI. 2000. Hitchhiking Under Positive Darwinian Selection.155(3): 1405-1413.
Fu W, O'connor TD, Jun G, Kang HM, Abecasis G, Leal SM, Gabriel SA, David, Shendure J, Nickerson DA, Others. 2013. Analysis of 6, 515 exomes reveals the recent origin of most human protein-coding variants.493(7431): 216-220.
Fu YX. 1995. Statistical properties of segregating sites.48(2): 172-197.
Fu YX, Li WH. 1993. Statistical tests of neutrality of mutations.133(3): 693-709.
Garud NR, Messer PW, Buzbas EO, Petrov DA. 2015. Recent selective sweeps in North American Drosophila melanogaster show signatures of soft sweeps.11(2): e1005004.
Gravel S, Henn BM, Gutenkunst RN, Indap AR, Marth GT, Clark AG, Yu F, Gibbs RA, Bustamante CD, Altshuler DL, Others. 2011. Demographic history and rare allele sharing among human populations.108(29): 11983-11988.
Griffiths RC, Tavaré S. 1998. The age of a mutation in a general coalescent tree.14(1-2): 273-295.
Griffiths RC. 1984. Asymptotic line-of-descent distributions.21(1): 67-75.
Griffiths RC, Tavare S. 1994. Simulating probability distributions in the coalescent.46(2): 131-159.
Gronau I, Hubisz MJ, Gulko B, Danko CG, Siepel A. 2011. Bayesian inference of ancient human demography from individual genome sequences.43(10): 1031-1034.
Grossman SR, Shylakhter I, Karlsson EK, Byrne EH, Morales S, Frieden G, Hostetter E, Angelino E, Garber M, Zuk O, Lander E S, Schaffner S F, Sabeti P C. 2010. A composite of multiple signals distinguishes causal variants in regions of positive selection.327(5967): 883-886.
Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD. 2009. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data.5(10): e1000695.
Han E, Sinsheimer JS, Novembre J. 2014. Characterizing bias in population genetic inferences from low-coverage sequencing data.31(3): 726-735.
Hancock AM, Witonsky DB, Ehler E, Alkorta-Aranburu G, Beall C, Gebremedhin A, Sukernik R, Utermann G, Pritchard J, Coop G, Di Rienzo A. 2010. Human adaptations to diet, subsistence, and ecoregion are due to subtle shifts in allele frequency.107(Supplement 2): 8924-8930.
Harris K, Nielsen R. 2013. Inferring demographic history from a spectrum of shared haplotype lengths.9(6): e1003521.
Hermisson J, Pennings PS. 2005. Soft sweeps: molecular population genetics of adaptation from standing genetic variation.169(4): 2335-2352.
Hobolth A, Christensen OF, Mailund T, Schierup MH. 2007. Genomic relationships and speciation times of human, chimpanzee, and gorilla inferred from a coalescent hidden Markov model.3(2): e7.
Huang XH, Kurata N, Wei XH, Wang ZX, Wang AH, Zhao Q, Zhao Y, Liu KY, Lu HY, Li WJ, Guo YL, Lu YQ, Zhou CC, Fan DL, Weng QJ, Zhu CR, Huang T, Zhang L, Wang YC, Feng L, Furuumi H, Kubo T, Miyabayashi T, Yuan XP, Xu Q, Dong GJ, Zhan QL, Li CY, Fujiyama A, Toyoda A, Lu TT, Feng Q, Qian Q, Li JY, Han B. 2012. A map of rice genome variation reveals the origin of cultivated rice.490(7421): 497-501.
Hufford MB, Xu X, Van Heerwaarden J, Pyhäjärvi T, Chia JM, Cartwright RA, Elshire RJ, Glaubitz JC, Guill KE, Kaeppler SM, Lai JS, Morrell PL, Shannon LM, Song C, Springer NM, Swanson-Wagner RA, Tiffin P, Wang J, Zhang GY, Doebley J, McMullen MD, Ware D, Buckler ES, Yang S, Ross-Ibarra J. 2012. Comparative population genomics of maize domestication and improvement.44(7): 808-811.
Jiang R, Tavaré S, Marjoram P. 2009. Population genetic inference from resequencing data.181(1): 187-197.
Jin L, Su B. 2000. Natives or immigrants: modern human origin in East Asia.1(2): 126-133.
Johnson PLF, Slatkin M. 2006. Inference of population genetic parameters in metagenomics: a clean look at messy data.16(10): 1320-1327.
Kamberov YG, Wang SJ, Tan JZ, Gerbault P, Wark A, Tan LZ, Yang YJ, Li SL, Tang K, Chen H, Powell A, Itan Y, Fuller D, Lohmueller J, Mao JH, Schachar A, Paymer M, Hostetter E, Byrne E, Burnett M, Mcmahon AP, Thomas MG, Lieberman DE, Jin L, Tabin CJ, Morgansend BA, Sabeti PC. 2013. Modeling recent human evolution in mice by expression of a selected EDAR variant.152(4): 691-702.
Ke YH, Su B, Song XF, Lu DR, Chen LF, Li HY, Qi CJ, Marzuki S, Deka R, Underhill P, Xiao CJ, Shriver M, Lell J, Wallace D, Wells RS, Seielstad M, Oefner P, Zhu DL, Jin JZ, Huang W, Chakraborty R, Chen Z, Jin L. 2001. African origin of modern humans in East Asia: a tale of 12, 000 Y chromosomes.292(5519): 1151-1153.
Keinan A, Mullikin JC, Patterson N, Reich D. 2007. Measurement of the human allele frequency spectrum demonstrates greater genetic drift in East Asians than in Europeans.39(10): 1251-1255.
Kim Y, Stephan W. 2002. Detecting a local signature of genetic hitchhiking along a recombining chromosome.160(2): 765-777.
Kimura M. 1955. Solution of a process of random genetic drift with a continuous model.41(3): 144-150.
Kong QP, Yao YG, Sun C, Bandelt HJ, Zhu CL, Zhang YP. 2003. Phylogeny of East Asian mitochondrial DNA lineages inferred from complete sequences.73(3): 671-676.
Lewontin RC, Krakauer J. 1973. Distribution of gene Frequency as a test of the Theory of the Selective Neutrality of Polymorphisms.74(1): 175-195.
Li HP, Stephan W. 2006. Inferring the demographic history and rate of adaptive substitution in Drosophila.2(10): e166.
Li H, Durbin R. 2011. Inference of human population history from individual whole-genome sequences.475(7357): 493-496.
Li H, Gu S, Cai XY, Speed WC, Pakstis AJ, Golub EI, Kidd JR, Kidd KK. 2008. Ethnic related selection for an ADH Class I variant within East Asia.3(4): e1881.
Li J, Zhang YP. 2009. Advances in research of the origin and domestication of domestic animals.17(4): 319-329. (in Chinese)
Linnen CR, Kingsley EP, Jensen JD, Hoekstra HE. 2009. On the origin and spread of an adaptive allele in deer mice.325(5944): 1095-1098.
Liu XM, Fu YX, Maxwell TJ, Boerwinkle E. 2010. Estimating population genetic parameters and comparing model goodness-of-fit using DNA sequences with error.20(1): 101-109.
Lohse K, Harrison RJ, Barton NH. 2011. A general method for calculating likelihoods under the coalescent process.189(3): 977-987.
Lukić S, Hey J. 2012. Demographic inference using spectral methods on SNP data, with an analysis of the human out-of-Africa expansion.192(2): 619-639.
Lukić S, Hey J, Chen K. 2011. Non-equilibrium allele frequency spectra via spectral methods.79(4): 203-219.
Lyu J, Zhang SL, Dong Y, He WM, Zhang J, Deng XN, Zhang YS, Li X, Li BY, Huang WQ, Wan WT, Yu Y, Li Q, Li J, Liu X, Wang B, Tao DY, Zhang GY, Wang J, Xu X, Hu FY, Wang W. 2013. Analysis of elite variety tag SNPs reveals an important allele in upland rice.4: 2138.
Lyu J, Li BY, He WM, Zhang SL, Gou ZH, Zhang J, Meng LY, Li X, Tao DY, Huang WQ, Hu FY, Wang W. 2014. A genomic perspective on the important genetic mechanisms of upland adaptation of rice.14(1): 160.
Mailund T, Dutheil JY, Hobolth A, Lunter G, Schierup MH. 2011. Estimating divergence time and ancestral effective population size of Bornean and Sumatran orangutan subspecies using a coalescent hidden Markov model.7(3): e1001319.
Mardis ER. 2008. The impact of next-generation sequencing technology on genetics.24(3): 133-140.
Marth GT, Czabarka E, Murvai J, Sherry ST. 2004. The allele frequency spectrum in genome-wide human variation data reveals signals of differeential demographic history in three large world populations.166(1): 351-372.
Nelson MR, Wegmann D, Ehm MG, Kessner D, Jean PS, Verzilli C, Shen JD, Tang ZZ, Bacanu SA, Fraser D, Warren L, Aponte J, Zawistowski M, Liu X, Zhang H, Zhang Y, Li J, Li Y, Li L, Woollard P, Topp S, Hall MD, Nangle K, Wang J, Abecasis G, Cardon LR, Zöllner S, Whittaker JC, Chissoe SL, Novembre J, Mooser V. 2012. An abundance of rare functional variants in 202 drug target genes sequenced in 14, 002 people.337(6090): 100-104.
Nielsen R, Williamson S, Kim Y, Hubisz MJ, Clark AG, Bustamante CD. 2005. Genomic scans for selective sweeps using SNP data.15(11): 1566-1575.
Ohta T, Kimura M. 1975. The effect of selected linked locus on heterozygosity of neutral alleles (the hitch-hiking effect).25(3): 313-326.
Patterson N, Price AL, Reich D. 2006. Population structure and eigenanalysis.2(12): e190.
Peng Y, Shi H, Qi XB, Xiao CJ, Zhong H, Ma RLZ, Su B. 2010. The ADH1B Arg47His polymorphism in East Asian populations and expansion of rice domestication in history.10(1): 15.
Peng Y, Yang ZH, Zhang H, Cui CY, Qi XB, Luo XJ, Tao X, Wu TY, Ouzhuluobu, Basang, Ciwangsangbu, Danzengduojie, Chen H, Shi H, Su B. 2011. Genetic variations in Tibetan populations and high-altitude adaptation at the Himalayas.28(2): 1075-1081.
Pritchard JK, Di Rienzo A. 2010. Adaptation-not by sweeps alone.11(10): 665-667.
Polanski A & Kimmel M. 2003. New explicit expressions for relative frequencies of single-nucleotide polymorphisms with application to statistical inference on population growth., 165(1): 427-436.
Pritchard JK, Pickrell JK, Coop G. 2010. The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation.20(4): R208-R215.
Przeworski M. 2003. Estimating the time since the fixation of a beneficial allele.164(4): 1667-1676.
Przeworski M, Coop G, Wall JD. 2005. The signature of positive selection on standing genetic variation.59(11): 2312-2323.
Qi JJ, Liu X, Shen D, Miao H, Xie BY, Li XX, Zeng P, Wang SH, Shang Y, Gu XF, Du YC, Li Y, Lin T, Yuan JH, Yang XY, Chen JF, Chen HM, Xiong XY, Huang K, Fei ZJ, Mao LY, Tian L, Städler T, Renner SS, Kamoun S, Lucas WJ, Zhang ZH, Huang SW. 2013a. A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity.45(12): 1510-1515.
Qi XB, Cui CY, Peng Y, Zhang XM, Yang ZH, Zhong H, Zhang H, Xiang K, Cao XY, Wang Y, Ouzhuluobu, Basang, Ciwangsangbu, Bianba, Gonggalanzi, Wu TY, Chen H, Shi H, Su B. 2013b. Genetic evidence of Paleolithic colonization and Neolithic expansion of modern humans on the Tibetan Plateau.30(8): 1761-1778.
Sabeti PC, Reich DE, Higgins JM, Levine HZP, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, Mcdonald GJ, Ackerman HC, Campbell SJ, Altshuler D, Cooper R, Kwiatkowski D, Ward R, Lander ES. 2002. Detecting recent positive selection in the human genome from haplotype structure.419(6909): 832-837.
Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R, Schaffner SF, Lander ES, International HapMap Consortium. 2007. Genome-wide detection and characterization of positive selection in human populations.449(7164): 913-918.
Schaffner SF, Foo C, Gabriel S, Reich D, Daly MJ, Altshuler D. 2005. Calibrating a Coalescent Simulation of Human Genome Sequence Variation.15(11): 1576-1583.
Schiffels S, Durbin R. 2014. Inferring human population size and separation history from multiple genome sequences.46(8): 919-925.
Sheehan S, Harris K, Song YS. 2013. Estimating variable effective population sizes from multiple genomes: A sequentially Markov conditional sampling distribution approach. Genetics, 194(3): 647-662.
Shi H, Su B. 2011. Molecular adaptation of modern human populations.2011: Article ID 484769.
Shi H, Tan SJ, Zhong H, Hu WW, Levine A, Xiao CJ, Peng Y, Qi XB, Shou WH, Ma RLZ, Li Y, Su B, Lu X. 2009. Winter temperature and UV are tightly linked to genetic changes in the p53 tumor suppressor pathway in Eastern Asia.84(4): 534-541.
Shriver MD, Kennedy GC, Parra EJ, Lawson HA, Sonpar V, Huang J, Akey JM, Jones KW. 2004. The genomic distribution of population substructure in four populations using 8, 525 autosomal SNPs.1(4): 274-286.
Simonson TS, Yang YZ, Huff CD, Yun HX, Qin G, Witherspoon DJ, Bai ZZ, Lorenzo FR, Xing JC, Jorde LB, Prchal JT, Ge RL. 2010. Genetic evidence for high-altitude adaptation in Tibet.329(5987): 72-75.
Smith JM, Haigh J. 1974. The hitch-hiking effect of a favourable gene.23(1): 23-25.
Tajima F. 1989. Statistical methods for testing the neutral mutations hypothesis by DNA polymorphism.123(3): 585-595.
Tang K, Thornton KR, Stoneking M. 2007. A new approach for using genome scans to detect recent positive selection in the human genome.5(7): e171.
Underhill PA, Passarino G, Lin AA, Shen P, Lahr MM, Foley RA, Oefner PJ, Cavalli-Sforza LL. 2001. The phylogeography of Y chromosome binary haplotypes and the origins of modern human populations.65(1): 43-62.
Voight BF, Kudaravalli S, Wen XQ, Pritchard JK. 2006. A map of recent positive selection in the human genome.4(3): e72.
Wang BB, Zhang YB, Zhang F, Lin HB, Wang XM, Wan N, Ye ZQ, Weng HY, Zhang LL, Li X, Yan JW, Wang PP, Wu TT, Cheng L, Wang J, Wang DM, Ma X, Yu J. 2011. On the origin of Tibetans and their genetic basis in adapting high-altitude environments.6(2): e17002.
Wang GD, Zhai WW, Yang HC, Fan RX, Cao X, Zhong L, Wang L, Liu F, Wu H, Cheng LG, Poyarkov AD, Poyarkov NA Jr, Tang SS, Zhao WM, Gao Y, Lv XM, Irwin DM, Savolainen P, Wu CI, Zhang YP. 2013. The genomics of selection in dogs and the parallel evolution between dogs and humans.4(5): 1860.
Weir BS, Cockerham CC. 1984. Estimating F-statistics for the analysis of population structure.38(6): 1358-1370.
Williamson SH, Hernandez R, Fledel-Alon A, Zhu L, Nielsen R, Bustamante CD. 2005. Simultaneous inference of selection and population growth from patterns of variation in the human genome.102(22): 7882-7887.
Wollstein A, Stephan W. 2015. Inferring positive selection in humans from genomic data.6(1): 5.
Wooding S, Rogers A. 2002. The matrix coalescent and an application to human single-nucleotide polymorphisms.161(4): 1641-1650.
Xia QY, Guo YR, Zhang Z, Li D, Xuan ZL, Li Z, Dai FY, Li YR, Cheng DJ, Li RQ, Cheng TC, Jiang T, Becquet C, Xu X, Liu C, Zha XF, Fan W, Lin Y, Shen YH, Jiang L, Jensen J, Hellmann I, Tang S, Zhao P, Xu HF, Yu C, Zhang GJ, Li J, Cao JJ, Liu SP, He NJ, Zhou Y, Liu H, Zhao J, Ye C, Du ZH, Pan GQ, Zhao AC, Shao HJ, Zeng W, Wu P, Li CF, Pan MH, Li JJ, Yin XY, Li DW, Wang J, Zheng HS, Wang W, Zhang XQ, Li SG, Yang HM, Lu C, Nielsen R, Zhou ZY, Wang J, Xiang ZH, Wang J. 2009. Complete resequencing of 40 genomes reveals domestication events and genes in silkworm ().326(5951): 433-436.
Xiang K, Ouzhuluobu, Peng Y, Yang ZH, Zhang XM, Cui CY, Zhang H, Li M, Zhang YF, Bianba, Gonggalanzi, Basang, Ciwangsangbu, Wu TY, Chen H, Shi H, Qi XB, Su B. 2013. Identification of a Tibetan-specific mutation in the hypoxic geneand its contribution to high-altitude adaptation.30(8): 1889-1898.
Xu SH, Li SL, Yang YJ, Tan JZ, Lou HY, Jin WF, Yang L, Pan XD, Wang JC, Shen YP, Wu BL, Wang HY, Jin L. 2011a. A genome-wide search for signals of high-altitude adaptation in Tibetans.28(2): 1003-1011.
Xu X, Liu X, Ge S, Jensen JD, Hu FY, Li X, Dong Y, Gutenkunst RN, Fang L, Huang L, Li JX, He WM, Zhang GJ, Zheng XM, Zhang FM, Li YR, Yu C, Kristiansen K, Zhang XQ, Wang J, Wright M, McCouch S, Nielsen R, Wang J, Wang W. 2011b. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes.30(1): 105-111.
Yao YG, Kong QP, Bandelt HJ, Rgen, Kivisild T, Zhang YP. 2002. Phylogeographic differentiation of mitochondrial DNA in Han Chinese.70(3): 635-651.
Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZXP, Pool JE, Xu X, Jiang H, Vinckenbosch N, Korneliussen TS, Zheng HC, Liu T, He WM, Li K, Luo RB, Nie XF, Wu HL, Zhao MR, Cao HZ, Zou J, Shan Y, Li SZ, Yang Q, Asan, Ni PX, Tian G, Xu JM, Liu X, Jiang T, Wu RH, Zhou GY, Tang MF, Qin JJ, Wang T, Feng SJ, Li GH, Huasang, Luosang J, Wang W, Chen F, Wang YD, Zheng XG, Li Z, Bianba Z, Yang G, Wang XP, Tang SH, Gao GY, Chen Y, Luo Z, Gusang L, Cao Z, Zhang QH, Ouyang WH, Ren XL, Liang HQ, Zheng HS, Huang YB, Li JX, Bolund L, Kristiansen K, Li YR, Zhang Y, Zhang XQ, Li RQ, Li SG, Yang HM, Nielsen R, Wang J, Wang J. 2010. Sequencing of 50 human exomes reveals adaptation to high altitude.329(5987): 75-78.
Zhang XM, Qi XB, Yang ZH, Serey B, Sovannary T, Bunnath L, Aun HS, Samnom H, Zhang H, Lin Q, van Oven M, Shi H, Su B. 2013. Analysis of mitochondrial genome diversity identifies new and ancient maternal lineages in Cambodian aborigines.4: 2599.
Zhao M, Kong QP, Wang HW, Peng MS, Xie XD, Wang WZ, Jiayang, Duan JG, Cai MC, Zhao SN, Cidanpingcuo, Tu YQ, Wu SF, Yao YG, Bandelt HJ, Zhang YP. 2009. Mitochondrial genome evidence reveals successful Late Paleolithic settlement on the Tibetan Plateau.106(50): 21230-21235.
Zhou ZK, Jiang Y, Wang Z, Gou ZH, Lyu J, Li WY, Yu YJ, Shu LP, Zhao YJ, Ma YM, Fang C, Shen YT, Liu TF, Li CC, Li Q, Wu M, Wang M, Wu YS, Dong Y, Wan WT, Wang X, Ding ZL, Gao YD, Xiang H, Zhu BG, Lee SH, Wang W, Tian ZX. 2015. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean.33(4): 408-414.
Received: 20 April 2015; Accepted: 19 June 2015
, E-mail: chenh@big.ac.cn
10.13918/j.issn.2095-8137.2015.4.223
杂志排行

Zoological Research的其它文章
- Extraction and identification of membrane proteins from black widow spider eggs
- Effects of frugivorous birds on seed retention time and germination in Xishuangbanna, southwest China
- The breeding biology of Red-Whiskered Bulbul (Pycnonotus jocosus) in Xishuangbanna, southwest China
- Why do we study animal toxins?
- Zoological Research is recognized as a core journal by the Research Center of Chinese Science Evaluation (RCCSE)
- The internationalization voyage of Zoological Research