锗硅烯与CH3OH加成反应机理及区域选择性
2015-12-05曾小兰
曾小兰 王 岩
(信阳师范学院化学化工学院, 河南 信阳 464000)
锗硅烯与CH3OH加成反应机理及区域选择性
曾小兰 王 岩*
(信阳师范学院化学化工学院, 河南 信阳 464000)
采用密度泛函理论方法, 在B3LYP/6-311++G(d,p)水平, 研究了几种锗硅烯与CH3OH的加成反应的微观机理和势能剖面, 分析了锗硅烯中Si=Ge双键的极性对加成反应区域选择性的影响. 研究结果表明, 锗硅烯可分别与CH3OH的单聚体或二聚体发生加成反应. 所有加成反应均从初始亲核或亲电复合物的形成开始. 母体锗硅烯H2Si=GeH2与CH3OH二聚体的加成反应比其与CH3OH单聚体的相应反应在动力学上更容易些, 但在其它锗硅烯与CH3OH的反应中情况则相反. 用Ph或SiMe3基团取代H2Si=GeH2中的H原子在动力学上使反应变得不利且SiMe3基团的影响更显著. 加成反应的区域选择性与锗硅烯中Si=Ge双键的极性以及Si-O (Ge-H)和Ge-O (Si-H)键的相对强弱都有关.
锗硅烯; 加成反应; 反应机理; 密度泛函理论; 区域选择性
1 引 言
与C原子处在同族的Si、Ge、Sn、Pb等元素形成的稳定不饱和化合物是主族元素化学的研究热点之一. 早在1981年, West等1成功地制备得到第一个包含Si=Si双键的稳定化合物. 自那以后, 很多包含M=M (M = Si, Ge, Sn, Pb)双键的重烯烃被合成得到, 它们的特殊结构和性质也逐渐为化学家所知.然而, 由两种不同元素形成的杂核重烯烃 >M=M'< (M, M' = Si, Ge, Sn, Pb)则相对较少. 1991年, Baines等2合成了第一种包含Si=Ge双键的锗硅烯化合物Mes2Si=GeMes2(Mes = C6H2-2,4,6-Me3). 此后,又陆续合成了几种锗硅烯, 并且测定了其中一些的单晶结构.3–10理论上, Grev等11,12利用从头算方法研究了母体锗硅烯H2Si=GeH2及其异构体的键解离能及相对稳定性. 实验测定和理论计算结果都表明锗硅烯一般为反式弯曲结构.
实验研究发现, 锗硅烯中的Si=Ge双键具有很高的反应活性, 其中研究较多的是它们与CH3OH间的1,2-加成反应. 实验结果表明, 当Mes2Si=GeMes2(1),2(tBu2MeSi)2Si=GeMes2(2)5或(tBuMe2Si)2Si=Ge(SiMe2tBu)2(3)10等三种锗硅烯分别在甲苯、四氢呋喃或正己烷溶剂中与CH3OH反应时均表现出完全的区域选择性. 在1及3与CH3OH的反应中, 只形成包含Si-O键的加成产物; 然而, 在涉及2的加成反应中, 则只形成包含Ge-O键的产物. 相关研究者推测上述结果的出现很可能是由锗硅烯中Si=Ge双键的极性所导致.
重烯烃特别是二硅烯与亲核试剂间1,2-加成反应的微观机理也是理论化学的研究热点之一. 1998年, Apeloig和Nakash13采用从头算(ab initio)方法研究了Me2Si=SiMe2与CH3OH及CF3OH的加成反应的机理. 随后, Kira研究组14–16采用从头算及密度泛函理论(DFT)方法对二硅烯与H2O及CH3OH的加成反应进行了系统的理论研究. 2008年, Yamabe等17采用DFT方法研究了几种二硅烯与醇及酚的单聚体及二聚体的反应, 发现二聚体反应一般比单聚体反应更有利些. 最近, Li和Su18采用DFT方法研究了具有很大张力的重烯烃(R2M=MR2, M = C, Si, Ge, Sn, Pb)与CH3OH单聚体及二聚体的反应, 发现涉及CH3OH二聚体的反应更为有利. 然而, 迄今为止, 有关锗硅烯与亲核试剂间的加成反应的理论研究尚未见报道. 本文采用DFT方法研究了几种锗硅烯与CH3OH间的1,2-加成反应的微观机理和势能剖面,比较了CH3OH单聚体及二聚体分别作为加成试剂时反应势能剖面的差别, 探讨了影响反应区域选择性的主要原因, 以期对锗硅烯的反应性有更深入的了解. 本文所研究的反应见图1.
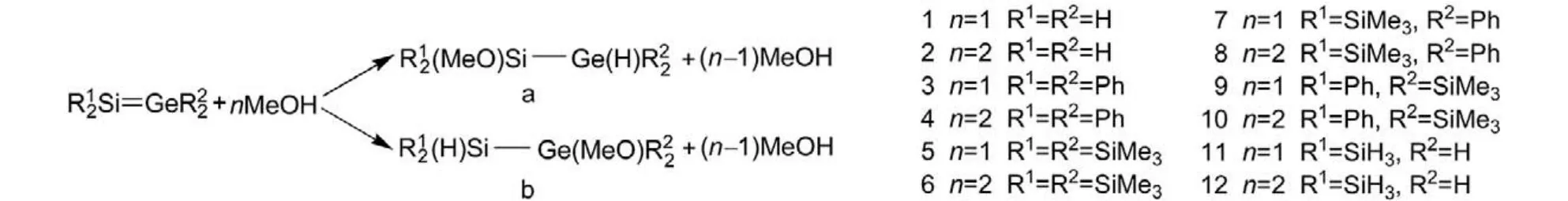
图1 锗硅烯与CH3OH的加成反应Fig.1 Addition reactions of germasilenes with CH3OH
2 计算方法
采用B3LYP/6-311++G(d,p)方法,19,20对反应中涉及的所有反应物、中间复合物、过渡态及产物的几何结构进行了全面优化. 通过频率分析计算,确认所得到的构型为无虚频的能量极小值点或具有一个虚频的过渡态. 根据过渡态的虚频振动模式判断它们位于正确的反应途径上. 对部分过渡态还进行了内禀反应坐标(IRC)计算以进一步确认它们确实连接相关的两个极小值. 考虑到溶剂的影响,采用极化连续反应场模型(PCM),21以甲苯(介电常数为2.37)或四氢呋喃(介电常数为7.43)为溶剂, 对部分反应进行B3LYP-SCRF/6-311++G(d,p)优化计算. 利用自然键轨道(NBO)方法,22,23求得各原子的电荷. 所有计算均采用Gaussian 09程序.24
3 结果与讨论
从锗硅烯与CH3OH的反应式(见图1)可以看出,本文所研究的加成反应共有24个. 为方便起见, 用符号Nx (x = a, b)表示这些反应, 其中的N表示反应序数, a表示形成一个Si-O键和一个Ge-H键的加成反应, b表示形成一个Ge-O键和一个Si-H键的反应, 这些反应中的复合物、过渡态和产物分别用CNx、TNx及PNx来表示.
对本文中所涉及的几种锗硅烯, 在B3LYP/6-311++G(d,p)水平优化得到的Si=Ge双键的键长在0.2229–0.2266 nm之间, 而根据文献报道,4,7,9,10从X射线衍射获得的几种锗硅烯的晶体结构中Si=Ge双键的键长在0.2221–0.2277 nm之间. 可以看出, Si=Ge双键键长计算值与实验测定值符合得很好,说明本文使用的理论方法是合理的.
3.1 非取代的锗硅烯与CH3OH的反应
3.1.1 反应机理
计算结果表明, 非取代的锗硅烯H2Si=GeH2(R1)与CH3OH的加成反应有两种可能的机理: R1与一分子或二分子CH3OH分别发生反应, 即图1中的反应1及反应2. 这些反应可分别看作是R1与CH3OH的单聚体或二聚体间的反应. 几何构型优化结果表明, R1与CH3OH单聚体形成Si-O键的反应1a及形成Ge-O键的反应1b都有两条反应途径, 即一个亲核途径和一个亲电途径. 在每个途径中, 两反应物分子先形成初始亲核或亲电复合物, 然后再经过两个过渡态和另一复合物形成最终产物. 两条不同途径的第二个复合物和第二个过渡态完全相同. 反应1a及1b的总的机理与文献报道的二硅烯与CH3OH单聚体间的加成反应14,15比较相似. 图2给出了反应1a及1b中复合物、过渡态及产物的分子结构示意图及与反应有关的四个原子的原子编号, 一些相关键的键长(nm)也列入其中.
从图2可以看出, 对反应1a的亲核途径, 在初始亲核复合物(C1a-1N(后缀N表示亲核途径, 下同))中, Si1-O2间距离仅比最终产物(P1a)中的Si-O单键长0.0261 nm, 说明Si1-O2间存在较强的相互作用. 从C1a-1N中两反应物分子的相对取向看, C1a-1N中存在O原子的孤对电子与R1的最低空轨道(LUMO)间的相互作用. NBO分析表明, 在C1a-1N的形成过程中有0.146e电荷从CH3OH分子转移到R1分子, 进一步说明C1a-1N为亲核型复合物. 图3给出了R1在B3LYP/6-311++G(d,p)水平计算得到的前线分子轨道示意图. 从图3可以看出, 由于R1分子具有反式弯曲构型, 其LUMO轨道的最大伸展方向与Si=Ge双键呈大于90°的角. 在C1a-1N中O2Si1Ge4键角大约为98°.
C1a-1N形成后, 接下来的反应步骤是经过第一个过渡态(T1a-1N)形成第二个复合物(C1a-2). 在该过程中, 整个分子的几何构型发生的主要变化是GeH2基团绕Si1-Ge4轴旋转大约180°. 在T1a-1N中, GeH2基团绕Si1-Ge4轴旋转了大约90°. C1a-2中的H3-Ge4距离(0.2588 nm)仍远大于P1a中的Ge-H单键(0.1537 nm), 说明在C1a-2中H3-Ge4间还只有很弱的相互作用. 接下来, C1a-2经过四元环过渡态(T1a-2)形成P1a, 主要的变化是O2-H3键的断裂和H3-Ge4键的形成, 同时还伴随有Si1-O2键的进一步形成. T1a-2的一个重要特征是其中的Si1、O2、H3及Ge4四个原子几乎在同一个平面上(Si1O2H3Ge4二面角为–4.68°).
在反应1a的亲电途径中, 两反应物分子首先形成初始亲电复合物C1a-1E(后缀E表示亲电途径, 下同), 其中的Si1-H3和Ge4-H3距离分别为0.3428及0.2946 nm, 说明C1a-1E中CH3OH与R1间的相互作用很弱. C1a-1E中两反应物分子的取向说明其中存在CH3OH的羟基H原子与R1的最高占据轨道(HOMO)间的亲电相互作用. NBO分析表明, 在C1a-1E中CH3OH分子片带有很少量的负电荷(–0.005e), 进一步证实C1a-1E为亲电型复合物. C1a-1E形成后, 接下来的反应步骤是O2原子对Si1的亲核进攻. 具体来说, CH3OH分子片绕Ge4-H3轴旋转以便O2原子的孤对电子接近Si1原子, 经过渡态(T1a-1E)形成与亲核途径相同的第二个复合物C1a-2.
反应1b的两条途径与反应1a非常相似. 从初始复合物经过第一个过渡态到第二个复合物, 主要的变化是Ge-O键的形成, 而从第二个复合物经过第二个过渡态到最终产物, 主要的变化是H-Si键的形成和O-H键的断开.
R1与CH3OH二聚体间的两个反应(2a及2b)的机理基本类似于1a和1b, 但只能优化得到亲核反应途径的分子间复合物和过渡态(见图2). 在反应物和最终产物之间可以优化出三个复合物和两个过渡态,其中前两个复合物及两个过渡态的分子几何结构与1a和1b的非常相似. 第三个复合物(C2a-3或C2b-3, 图2未列出)实际上是P1a或P1b与一个CH3OH分子通过O-HO氢键相互作用形成的. 类似于四元环过渡态T1a-2及T1b-2, 六中心过渡态T2a-2及T2b-2中的Si(Ge)1、O2、H3、O4、H5及Ge(Si)6六个原子几乎在同一个平面上.
3.1.2 势能剖面
以反应物分子的总能量及298.15 K时Gibbs自由能之和作为能量零点计算得到了反应1a、1b、2a及2b所涉及的分子间复合物、过渡态及产物的相对能量(Er)及相对Gibbs自由能(Gr). 图4给出了这些反应的自由能势能图. 对反应2a及2b, 考虑到CH3OH单聚体与二聚体之间可能存在的平衡, 选择R1与两个孤立的CH3OH分子的能量或Gibbs自由能之和作为能量零点.
从图1可以看出, 反应1a和2a的产物的Gr相同,说明1a和2a在热力学上是完全等价的, 1b和2b也是如此. 但1a(1b)和2a(2b)的活化Gibbs自由能并不相同. 从图4可以发现, P1a和P1b的Gr分别为–179.3及–126.6 kJmol–1, 说明反应1a(2a)及1b(2b)室温下均能正向进行到底. 因此, 对R1与CH3OH单聚体或二聚体之间的加成反应, Si-O键和Ge-O键哪个较易形成由反应的活化Gibbs自由能的相对高低决定. 也就是说, 加成反应的区域选择性由动力学因素所决定. 另外, P1a和P1b具有较负的Gr值主要是由于P1a和P1b具有较负的Er值(–222.4及–169.3 kJmol–1)或者说主要是由于加成反应为强放热过程造成的, 这说明R1中的Si=Ge双键比较弱. 在涉及Si原子的其他双键体系中也有类似的情况.25–27

图2 一些复合物, 过渡态及产物的分子结构示意图Fig.2 Schematic diagrams of molecular structures of some complexes, transition states, and products

图3 H2Si=GeH2(R1)的HOMO与LUMO示意图Fig.3 Schemes for HOMO and LUMO of H2Si=GeH2(R1)
从图4可以看出, 反应1a和1b的六个分子间复合物及反应2b的前两个复合物的Gr为正值, 说明它们在室温下很难稳定存在. 由于这些复合物的Er均为负值, 上述结果显然是由负的熵因素造成的. 反应2a的第二个过渡态即T2a-2的Gr反而比C2a-2低2.8 kJmol–1, 这可能是因为C2a-2的零点能比T2a-2高的缘故. 实际上, T2a-2的Er比C2a-2高约4 kJmol–1.
比较反应1a、1b、2a及2b的两步反应过程的过渡态的Gr数值可以看出, 在多数情况下, 第一步反应过程即第二个复合物的形成为速率控制步骤. 但在反应1b的亲电反应途径中, 第二步反应过程即最终产物P1b的形成为速率控制步骤.
从图4可以看出, C1a-1N及T1a-1N的Gr(10.6和30.0 kJmol–1)分别低于C1a-1E及T1a-1E的Gr(22.5和36.4 kJmol–1), 说明反应1a主要通过亲核途径来进行. 而反应1b主要通过亲电途径来进行. 根据NBO分析(见表1), 在R1分子中, Si和Ge原子所带电荷分别为+0.293e和+0.204e. 由于Si原子带有相对更多的正电荷, 形成Si-O键的反应1a的亲核途径相对更有利些, 而形成Ge-O键的反应1b则相反.

图4 反应1a, 1b, 2a及2b的自由能势能面Fig.4 Free energy profiles of the reactions 1a, 1b, 2a, and 2b
比较反应1a(b)和2a(b)的第二个过渡态的Er及Gr数值可以发现, T2a-2或T2b-2的Er及Gr明显低于T1a-2或T1b-2的Er及Gr, 但Er的差别明显大于Gr的差别. 例如, T2a-2的Er比T1a-2低79.2 kJmol–1, 但其Gr只比T1a-2低34.9 kJmol–1. 这说明造成T2a-2(T2b-2)与T1a-2(T1b-2)之间的Gr的差别有两种相反的因素, 能量因素使得前者的Gr小于后者, 而熵因素的影响则相反. 显然, 能量因素的影响更显著些. 这是因为, T1a-2和T1b-2为具有较大张力的四元环过渡态且其中没有氢键存在, 而T2a-2及T2b-2是具有较小张力的六中心过渡态且其中存在O—HO氢键相互作用. 上述结果与文献报道的有机分子的水助分子内或分子间的质子转移过程28,29比较相似. 在文献报道的乙烯与HF的加成反应30及我们以前所研究的硅苯与HX (X = F, OH, NH2)的1,2-及1,4-加成反应31中也有类似的结果.
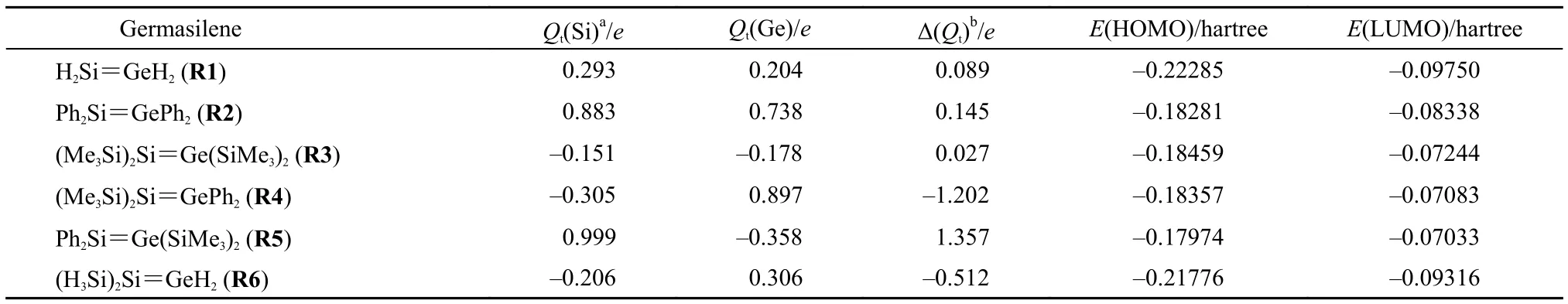
表1 锗硅烯中双键Si原子和Ge原子的NBO电荷及前线轨道(FMO)能级Table1 Natural bond orbital (NBO) atomic charges on the double bond Si and Ge atoms and the energy level of frontier molecular orbital (FMO) for germasilenes
比较反应 1a(b)和2a(b)的最有利的反应途径的过渡态的Gr数值可以看出, 当CH3OH与R1发生加成反应时, CH3OH二聚体比CH3OH单聚体更为活泼.这可能是因为, 与单聚体相比, CH3OH二聚体的末端H原子有相对较强的酸性, 而其中的参与反应的O原子有相对较强的碱性. NBO分析表明, 在CH3OH分子中, O原子以及羟基H原子的电荷分别为–0.729e和0.451e; 而在CH3OH二聚体分子中, 相应的电荷分别为–0.763e和0.465e.
从图4可以看出, R1与CH3OH间形成Si-O键的最有利的反应途径即反应2a的亲核反应途径中两个过渡态的Gr分别为6.0及–18.7 kJmol–1, 二者间形成Ge-O键的最有利的反应途径即反应2b的亲核反应途径的两个过渡态的Gr分别为46.1及23.9 kJmol–1.显然, 形成Si-O键的反应在动力学上远较形成Ge-O键的反应有利. 根据本文的计算结果, R1与CH3OH间的加成反应可表现出几乎完全的区域选择性.
3.2 Ph2Si=GePh2, (Me3Si)2Si=Ge(SiMe3)2, (Me3Si)2Si=GePh2, Ph2Si=Ge(SiMe3)2及(H3Si)2Si=GeH2与CH3OH的反应
实验研究表明, 三种锗硅烯(Mes2Si=GeMes2, (tBu2MeSi)2Si=GeMes2及(tBuMe2Si)2Si=Ge(SiMe2
tBu)2)与CH3OH发生加成反应都表现出完全的区域选择性, 即只能观察到形成Si-O键或Ge-O键的产物. 为探讨这些反应中区域选择性的起源, 本文还研究了几种取代的锗硅烯与CH3OH间的加成反应. 由于Mes, SiMe2tBu或SiMetBu2取代基包含的原子数目较多, 考虑到计算条件, 其中的Mes取代基用苯基代替, 而SiMe2tBu或SiMetBu2取代基用SiMe3代替. 本节考虑的几种锗硅烯为Ph2Si=GePh2(R2), (Me3Si)2Si=Ge(SiMe3)2(R3), (Me3Si)2Si=GePh2(R4)及Ph2Si=Ge(SiMe3)2(R5). 此外, 还研究了(H3Si)2Si=GeH2(R6)与CH3OH的加成反应.
3.2.1 反应机理
从表1可以看出, 由于Ph基团具有较强的吸电子性质, 使得R2中的Si和Ge原子比R1中均带有更多的正电荷(0.883e及0.738e), 但仍然是Si原子带有较多的正电荷. 由于SiMe3为较强的给电子基团, 使得R3中的Si和Ge原子均带有少量的负电荷(–0.151e及–0.178e), 但Ge原子带有相对更多的负电荷. 从R1、R2及R3中Si和Ge原子的电荷差别来看, 这三种锗硅烯中Si=Ge双键的极性大致相当. 由于R4及R5中的双键Si原子和Ge原子携带具有相反性质的基团, 使得其中的Si=Ge双键都有很强的极性但极性正好相反. R6中的双键Si原子携带给电子基团SiH3, 使得其中的Si=Ge双键的极性与R4性质相同, 但极性相对较弱. 比较本文涉及的几种锗硅烯的FMOs能级可以看出, 除R1以外的其它锗硅烯的HOMO和LUMO能级都比R1的高, 预示这些锗硅烯可能更容易发生亲电加成反应.
计算结果表明, R2与CH3OH间的加成反应(图1中的反应3和反应4)的机理与反应1非常相似. 无论是形成Si-O键还是Ge-O键的反应, 无论是与CH3OH单聚体还是与CH3OH二聚体的反应, 都有亲核和亲电两条途径. 对R3与CH3OH间的四个反应5a、5b、6a及6b, 都未能优化得到初始亲核复合物及随后的过渡态, 说明这四个反应都不存在亲核反应途径, 这可能是因为R3中Si和Ge原子均带负电荷且R3有比R1和 R2更高的LUMO能级. 在R4及R5与CH3OH间的八个加成反应中, 情况较复杂. 其中的部分反应有亲核和亲电两个途径, 而另一些反应只存在亲电反应途径. 另外, 还可发现, 反应5b、7b及9b的亲电反应途径与其它亲电反应不同. 在这三个反应中, 只能优化出初始亲电复合物和第二个(四元环)过渡态, 说明它们为一步亲电加成反应. 该机理与文献报道的二硅烯与HF及HCl的加成反应32的情况很相似.
3.2.2 反应的势能剖面
图S1–S5 (见Supporting Information)给出了本节所讨论的各反应的自由能势能图. 对R2与CH3OH间的加成反应(3a、3b、4a及4b), 我们还计算得到了反应途径上各驻点在甲苯溶剂中的Gr数值. 对R4与CH3OH间的加成反应(7a、7b、8a及8b),还计算得到了各驻点在四氢呋喃溶剂中的Gr数值.比较这些反应在气相和甲苯(四氢呋喃)溶剂中的Gr数值可以发现, 甲苯溶剂对加成反应的势能剖面的影响很小, 溶剂效应引起的Gr的改变一般不超过10 kJmol–1. 四氢呋喃溶剂的影响相对较大, 且一般使加成反应变得不利. 这可能是因为在反应7a、7b、8a及8b中, 产物分子的极性小于反应物分子, 具有较强极性的四氢呋喃溶剂对反应物分子的稳定化作用相对更显著一些. 但四氢呋喃溶剂对形成Si-O键和Ge-O键的反应的影响大致相同, 说明四氢呋喃溶剂并不改变反应的区域选择性. 因此, 下面的讨论主要基于气相中的结果.
计算结果表明, 在R2、R3、R4及R5与CH3OH间的所有加成反应中, 产物的Gr均为很大的负值(–79.5 – –181.5 kJmol–1), 说明这些反应的区域选择性同样由动力学因素决定. 比较各反应中产物的Gr数值可发现, P3a或P3b的Gr(–181.5及–128.0 kJmol–1)与P1a或P1b的非常接近, 说明用四个Ph取代R1中的H原子对反应的热力学性质基本无影响.但P5a或P5b的Gr分别比P1a或P1b高大约55或36 kJmol–1, 说明SiMe3取代基在热力学上对反应非常不利, 显然, 这是由于SiMe3有很大的空间位阻效应造成的. P7a的Gr比P1a高约60 kJmol–1, 而P7b的Gr只比P1b高约13 kJmol–1, 也是基于同样的原因. 另外, SiMe3有比Ph大得多的空间位阻效应也体现在其使过渡态的Gr数值有更为显著的增加.
比较同一反应的亲核及亲电途径的过渡态的Gr可以发现, 在部分反应中(如3a, 4a和8a), 亲核途径和亲电途径哪种在动力学上更有利与锗硅烯中Si=Ge双键的极性相符合. 但在另外一些反应中却出现了不一致的结果, 这很可能是由于SiMe3有很强的空间位阻效应造成的. 反应7b只存在亲电途径可能也与此有关.
从图S1–S5可以看出, 对于本节讨论的加成反应, 若反应途径中包含两个过渡态, 在绝大多数情况下, 第一步过程即第二个复合物的形成为速率控制步骤. 比较R2、R3、R4或R5与CH3OH单聚体及二聚体间的反应(以下分别简称为单聚体反应或二聚体反应)中相应过渡态的Gr数值可以看出, 二聚体反应的第二个(六中心)过渡态的Gr总是比相应单聚体反应的第二个(四中心)过渡态的低. 若二聚体反应和单聚体反应都有相同类型(亲核或亲电)的两步反应过程, 如反应5a和6a等, 可以发现, 前者的第一个过渡态的Gr都比后者的相应过渡态的高, 这可能是由于在二聚体反应的第一步过程中空间位阻效应更显著所导致. 由于第一步过程一般为速率控制步骤, 当二聚体反应和单聚体反应都有相同类型的两步反应过程时, 前者在动力学上一般不如后者有利, 本节讨论的反应多数情况下是这样. 但若二聚体反应和单聚体反应的机理不完全相同时, 情况则比较复杂. 例如, 8b比7b有利, 但10b不如9b有利. 对反应5b和6b, 由于未能优化得到6b中前两个复合物之间的过渡态(可能是因为该途径的势能剖面比较平缓造成的), 无法判断5b和6b中哪一个更有利些.综合以上结果, 对本节讨论的R2、R3、R4或R5与 CH3OH间的反应, 多数情况下, 二聚体反应不如单聚体反应有利.
比较本节讨论的各反应中形成Si-O键及Ge-O键的最有利的反应途径中各过渡态的Gr数值可以看出, 在R2及R3作为反应物的反应中, Si-O键的形成在动力学上比Ge-O键的形成有利.例如, T3a-1N及T3a-2的Gr分别比T3b-1E及T3b-2低19.0或35.3 kJmol–1. 本文的计算结果预计这些反应有明显的区域选择性, 这与前面提到的Mes2Si=GeMes2及(tBuMe2Si)2Si=Ge(SiMe2tBu)2与CH3OH的加成反应的实验结果完全一致. 在涉及R5的反应中, 同样也是形成Si-O键的反应在动力学上更有利, 但T9a-2(T10a-2)与T9b-E(T10b-2)之间的Gr差别明显比R2及R3作为反应物时两个竞争反应的第二个(四元或六中心)过渡态的Gr差别更大些. 另一方面, 在涉及R4的反应中, 情况与上面恰好相反, Ge-O键的形成在动力学上远比形成Si-O键有利,说明 R4与CH3OH间的加成反应也呈现出明显的区域选择性, 只不过与上面三种情况及涉及R1的反应刚好相反. 此结果与涉及(tBu2MeSi)2Si=GeMes2的加成反应的实验观察一致. 从表1可以看出, R1、R2、R3及R5中Si=Ge双键的极性都是一致的, 其中的Si原子带有较多的正电荷或较少的负电荷, 而R4中的Si=Ge双键则具有颠倒的极性. 上述结果表明, 当锗硅烯与CH3OH发生加成反应时, 反应的区域选择性与锗硅烯中Si=Ge双键的极性有明显的相关性, CH3OH中的O原子优先进攻Si=Ge双键中携带较多正电荷或较少负电荷的原子, 这与通常的化学直观一致. 然而, 应当注意的是, R1、R2及R3中的Si=Ge双键虽然有一定的极性, 但极性都比较弱, 说明实验观察到的区域选择性可能并不是仅由Si=Ge双键的极性所决定. 为此, 我们还用同样方法研究了(H3Si)2Si=GeH2(R6)与CH3OH的加成反应(图1中的反应11a、11b、12a及12b). R6中的双键Si原子和Ge原子所带电荷分别为–0.206e及0.306e.根据上面的结果, 若加成反应的区域选择性仅由Si=Ge双键的极性所决定, 则当R6与CH3OH发生加成反应时, Ge-O键的形成应当更有利. 计算结果表明, 形成Si-O键的最有利的反应途径中两个过渡态的Gr分别为39.0及12.9 kJmol–1, 而形成Ge-O键的对应Gr分别为32.8及58.7 kJmol–1, 说明Si-O键的形成实际更有利. 我们认为, 锗硅烯与CH3OH间加成反应的区域选择性不仅与Si=Ge双键的极性密切相关, 而且与Si-O (Ge-H)及Ge-O (Si-H)键的相对强弱有关. 根据上面的讨论, 对R1与CH3OH间的加成反应, P1a的Gr比P1b低大约53 kJmol–1, 且该差别主要由能量因素即Er的差别所决定. 此结果说明, 单纯从键能的角度看, Si-O (Ge-H)键的形成比Ge-O (Si-H)键有利. 因此, 锗硅烯与CH3OH间的加成反应可分为三种情况: (1)若其中的Si=Ge双键有正常的极性(Si原子带有相对更多的正电荷), 如R1、R2、R3及R5中, 无论极性强弱如何, 都是Si-O (Ge-H)键的形成更有利; (2) 若Si=Ge双键有颠倒的极性但极性不太强时, 如R6中, 键能的影响起主要作用, 仍然是Si-O (Ge-H)键的形成更有利; (3) 当Si=Ge双键有颠倒的极性且极性很强时, 如R4中, 极性的影响起主要作用, 此时, Ge-O (Si-H)键的形成更有利. 另外,当锗硅烯中的Si原子或Ge原子连有体积庞大的取代基时, 空间位阻效应对加成反应的区域选择性也可能有影响.
4 结 论
采用DFT-B3LYP/6-311++G(d,p)方法研究了锗硅烯与CH3OH单聚体及二聚体间的加成反应的微观机理、势能剖面及区域选择性, 得到以下结论.大部分反应为两步反应过程, 其中的第一步主要是Si-O键或Ge-O键的形成, 而第二步主要是Ge-H键或Si-H键的形成. 同一反应同时存在亲核和亲电两种反应途径时, 哪种在动力学上更有利与电子效应及空间位阻效应都有关. 当CH3OH与母体锗硅烯H2Si=GeH2发生加成反应时, CH3OH二聚体比CH3OH单聚体更为活泼; 但若锗硅烯中的Si原子和Ge原子上存在体积较大的取代基时, 单聚体反应一般更有利些. 锗硅烯中Si(Ge)原子上的Ph或SiMe3取代基在动力学上不利于反应的进行且SiMe3的影响更为显著. 加成反应的区域选择性由动力学因素控制. 甲苯溶剂对加成反应的影响较小,而四氢呋喃溶剂对加成反应明显不利.
Supporting Information: The free energy profiles of the reactions 3a(b)–12a(b) have been included. This information is available free of charge via the internet at http://www.whxb.pku. edu.cn.
(1)West, R.; Fink, M. J.; Michl, J. Science 1981, 214, 1343. doi: 10.1126/science.214.4527.1343
(2)Baines, K. M.; Cooke, J. A. Organometallics 1991, 10, 3419. doi: 10.1021/om00056a004
(3)Lee, V. Y.; Ichinohe, M.; Sekiguchi, A.; Takagi, N.; Nagase, S. J. Am. Chem. Soc. 2000, 122, 9034. doi: 10.1021/ja001551s
(4)Lee, V. Y.; Ichinohe, M.; Sekiguchi, A. J. Am. Chem. Soc. 2000, 122, 12604. doi: 10.1021/ja0030921
(5)Ichinohe, M.; Arai, Y.; Sekiguchi, A.; Takagi, N.; Nagase, S. Organometallics 2001, 20, 4141. doi: 10.1021/om010419d
(6)Sekiguchi, A.; Izumi, R.; Ihara, S.; Ichinohe, M.; Lee, V. Y. Angew. Chem. Int. Edit. 2002, 41, 1598. doi: 10.1002/1521-3773(20020503)41:9<1598::AID-ANIE1598>3.0.CO;2-8
(7)Iwamoto, T.; Masuda, H.; Kabuto, C.; Kira, M. Organometallics 2005, 24, 197. doi: 10.1021/om049183e
(8)Iwamoto, T.; Abe, T.; Kabuto, C.; Kira, M. Chem. Commun. 2005, 5190.
(9)Igarashi, M.; Ichinohe, M.; Sekiguchi, A. Heteroat. Chem. 2008, 19, 649. doi: 10.1002/hc.v19:7
(10)Iwamoto, T.; Okita, J.; Yoshida, N.; Kira, M. Silicon 2010, 2, 209. doi: 10.1007/s12633-011-9069-8
(11)Grev, R. S.; Schaefer, H. F., III; Baines, K. M. J. Am. Chem. Soc. 1990, 112, 9458. doi: 10.1021/ja00182a003
(12)Grev, R. S.; Schaefer, H. F., III. Organometallics 1992, 11, 3489. doi: 10.1021/om00059a002
(13)Apeloig, Y.; Nakash, M. Organometallics 1998, 17, 2307. doi: 10.1021/om980016m
(14)Takahashi, M.; Veszprémi, T.; Hajgató, B.; Kira, M. Organometallics 2001, 19, 4660. doi: 10.1021/om000385u
(15)Veszprémi, T.; Takahashi, M.; Hajgató, B.; Kira, M. J. Am. Chem. Soc. 2001, 123, 6629. doi: 10.1021/ja0040823
(16)Takahashi, M.; Veszprémi, T.; Kira, M. Organometallics 2004, 23, 5768. doi: 10.1021/om049418m
(17)Yamabe, S.; Mizukami, N.; Tsuchida, N.; Yamazaki, S. J. Organomet. Chem. 2008, 693, 1335. doi: 10.1016/ j.jorganchem.2008.01.035
(18)Li, B. Y.; Su, M. D. J. Phys. Chem. A 2012, 116, 4222. doi: 10.1021/jp3018138
(19)Becke, A. D. J. Chem. Phys. 1993, 98, 5648. doi: 10.1063/ 1.464913
(20)Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785. doi: 10.1103/PhysRevB.37.785
(21)Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Chem. Phys. Lett. 1996, 255, 327. doi: 10.1016/0009-2614(96)00349-1
(22)Reed, A. E.; Weinstock, R. B.; Weinhold, F. J. Chem. Phys. 1985, 83, 735. doi: 10.1063/1.449486
(23)Reed, A. E.; Weinhold, F. J. Chem. Phys. 1985, 83, 1736. doi: 10.1063/1.449360
(24)Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; et al. Gaussian 09, Revision A.02; Gaussian Inc.: Wallingford, CT, 2009.
(25)Wang, X.; Huang, Y.; An, K.; Fan, J.; Zhu, J. J. Organomet.Chem. 2014, 770, 146. doi: 10.1016/j.jorganchem.2014.08.018
(26)Wang, X.; Zhu, C.; Xia, H.; Zhu, J. Organometallics 2014, 33, 1845. doi: 10.1021/om500170w
(27)Huang, Y.; Zhu, J. Chem. Asian J. 2015, 10, 405. doi: 10.1002/asia.v10.2
(28)Zhou, Z. Y.; Liu, M.; Su, Z. M.; Xie, Y. Z.; Ding, S. D.; Wang, H. J. Acta Phys. -Chim. Sin. 2011, 27, 2035. [周子彦, 刘 敏, 苏忠民, 谢玉忠, 丁慎德, 王华静. 物理化学学报, 2011, 27, 2035.] doi: 10.3866/PKU.WHXB20110903
(29)Yang, G. H.; Li, Y. X.; Yan, S. H.; Dai, L.; Zhao, B. Acta Chim. Sin. 2011, 69, 1743. [杨国辉, 李言信, 颜世海, 代 丽, 赵 斌.化学学报, 2011, 69, 1743.]
(30)Clavero, C.; Duran, M.; Lledos, A.; Ventura, O. N.; Bertran, J. J. Am. Chem. Soc. 1986, 108, 923. doi: 10.1021/ja00265a014
(31)Wang, Y.; Zeng, X. L. Acta Phys. -Chim. Sin. 2012, 28, 2831. [王 岩, 曾小兰. 物理化学学报, 2012, 28, 2831.] doi: 10.3866/PKU.WHXB201208134
(32)Hajgató, B.; Takahashi, M.; Kira, M.; Veszprémi, T. Chem. -Eur. J. 2002, 8, 2126. doi: 10.1002/1521-3765(20020503)8:9 <2126::AID-CHEM2126>3.0.CO;2-2
Mechanism and Regioselectivity of Addition Reactions of CH3OH to Germasilenes
ZENG Xiao-Lan WANG Yan*
(College of Chemistry and Chemical Engineering, Xinyang Normal University, Xinyang 464000, Henan Province, P. R. China)
Density functional theory (DFT) calculations of the reaction mechanisms and potential energy surfaces for the addition reactions of CH3OH to several germasilenes were performed at the B3LYP/6-311++G(d,p) level. The effect of the polarity of the Si=Ge double bond in germasilenes on the regioselectivity of the addition reactions was also investigated. The results indicate that germasilenes can react with a monomer or dimer of CH3OH. All reactions start with formation of nucleophilic or electrophilic complexes. The dimer of CH3OH adds to H2Si=GeH2kinetically more easily than the monomer. However, the situation is generally the opposite for substituted germasilenes. There is a kinetic disadvantage of substituting phenyl (Ph) or SiMe3groups for H atoms in H2Si=GeH2in the addition reactions, and the effect of the SiMe3group is more remarkable than that of the Ph substituent. Both the polarity of the Si=Ge double bond and the strength of the Si-O (Ge-H) and Ge-O (Si-H) bonds affect the regioselectivity of the addition reactions.
Germasilene; Addition reaction; Reaction mechanism; Density functional theory; Regioselectivity
O641
10.3866/PKU.WHXB201507202
Received: May 7, 2015; Revised: July 20, 2015; Published on Web: July 20, 2015.
*Corresponding author. Email: wangyanxytc@163.com; Tel: +86-376-6390702.
The proejct was supported by Henan Provincial Fundamental and Frontier Technological Research Program, China (142300410194).
河南省基础与前沿技术研究计划(142300410194)资助项目
© Editorial office of Acta Physico-Chimica Sinica
