催化甾体羟基化的P450氧化酶BM3的蛋白质工程的研究进展
2015-11-27刘星孔建强
刘星,孔建强
·综述·
催化甾体羟基化的P450氧化酶BM3的蛋白质工程的研究进展
刘星,孔建强
甾体关键位点的羟基化在药用甾体的合成中发挥重要作用。为探究细胞色素 P450(CYP450)氧化酶 BM3 在甾体羟基化合成中的潜在应用,基于细胞色素 P450 BM3 的蛋白质工程逐渐发展起来。在取得的若干 P450 BM3 突变酶的基础上,通过新一代测序技术和生物信息学分析等方法,筛选出催化甾体羟基化的相关 CYP450。根据易错 PCR等定向进化技术获得了突变位点信息,进一步采用(饱和)定点突变等进化技术对活性氨基酸位点进行分析,再经过筛选获得既高于亲本酶也高于易错 PCR 技术得到的突变酶活力的新突变酶,并对突变体进行功能验证,进一步阐明甾体羟基化的可行性和重要性。此外,P450 BM3 催化底物和生成产物的选择性可以通过迭代的组合活性位点突变而改变。本文旨在探究近年来科研人员在 P450 氧化酶 BM3蛋白质工程催化甾体羟基化的改良领域中所做的尝试、获得的成果以及存在的问题,为 P450 BM3 羟基化疏水性甾体的深入研究提供理论依据。
1 概述
甾体药物在医药领域中占有重要地位,是仅次于抗生素的第二大类药物[1],具有很强的抗过敏、抗感染、抗病毒等药理活性。除了薯蓣皂苷元在甾体药物中的广泛应用外,一些甾醇如胆固醇、谷甾醇和菜籽甾醇等现在也被认为是具有巨大潜能的甾体激素类药物的起始原料。CYP450 酶在甾体激素生物合成及代谢清除过程中起关键作用,而甾体激素也可影响 P450 酶的表达及代谢活性[2]。
众所周知,甾体激素结构的微小变化,例如甾体母核的氧化状态,附加的官能团的类型、数量和立体位置[3]等会严重影响它们的生物活性。为了获得新颖的和更有活性的化合物,人们对甾类化合物进行了大量研究。在这些研究中,羟基化引起了人们越来越高的关注。羟基化增加了疏水性类甾体的极性,且羟基化的甾体通常比非羟基化的甾体具有更高的生物活性。甾体的羟基化主要是运用微生物全细胞催化法[3],只有少数是使用分离的酶。细胞色素 P450 单加氧酶超家族是能催化甾体羟基化酶的典型代表,它们具有反应条件温和、环境友好、催化效率和选择性高等优势,能完全满足化学合成的要求[4-5]。
甾体关键部位的羟基化提供了一个有效的获得珍贵药物代谢物、天然产物的衍生物和其他重要化合物的途径[6]。然而,由于 P450 酶的内在特性[7],如需要合适的电子转移系统等,还不能进行大规模工业应用,但通过蛋白质工程得到的细胞色素 P450 BM3 及其突变体已经被报道能在不同的位置羟基化甾体。
1.1 CYP450
CYP450 是一类含亚铁血红素的单加氧酶,广泛存在于各类生物体内,参与多种外源物质的代谢和内源物质的转化,如甾类激素、胆汁酸、胆固醇等的代谢。而在人类药物代谢中起决定作用的主要是 CYP1、CYP2 以及 CYP3 酶亚家族[8]。尽管 CYP450 氨基酸序列的不一致性高达 90%,但是其序列拥有共同的折叠方式[9]。CYP450 酶能够催化的反应主要有烃类化合物的羟基化或环氧化,杂环化合物的氨、氧、硫部位上的脱烷基化等。在真核生物中,这些酶对类固醇的合成和生物转化起着重要作用。而哺乳动物肝脏的CYP450 对许多药物发挥重要的解毒作用[10]。虽然这些酶在自然进化中还没有具备现代化学工业所需的催化特性,许多CYP450 显示出宽松的底物特异性,表现为对非天然底物具有一定程度的活性,如应用于一些药物的开发[11]。
多数 P450 单加氧酶以膜结合形式存在,其发挥活性需要依赖复杂的电子转移系统和昂贵的辅因子,如 NADPH,往往活性较低,且稳定性也较差。而 CYP102 家族的细菌性 P450 单加氧酶却可以克服上述部分难题。例如,来源于巨大芽孢杆菌(Bacillus Megaterium)的 BM3,它是已知的最活跃的一种含血红素的 CYP450。含血红素的 P450 单加氧酶可构成一个主要的酶家族,其含有 4500 多个来源于不同物种的成员[12],并且各成员均以可溶性形式存在,同时具有较高的转化率,推测这可能是由于其 N-末端的血红素加氧酶结构域和 C 末端的黄素还原酶结构域共价连接所致。此外,含血红素的 P450 单加氧酶家族的成员还具有独一无二的功能,即把羟基引入底物的非活性碳上。这一功能具有重要意义,因为在一个特定的底物插入羟基可以显著影响其生物利用度、生物活性和溶解度[6]。基于制药行业和生物技术的需求,目前甾体是科研人员最感兴趣的底物之一。尽管 CYP450 的种类不同,所需的电子传递系统也不尽相同,但一般可以用以下通式来表示其催化反应:
RH + O2+ NADPH + H+→ ROH + NADP++ H2O
式中 RH 代表底物,在分子氧(O2)参与下底物被 P450催化形成水和羟基化产物(ROH)。其氧化机制见图1[13-14]。
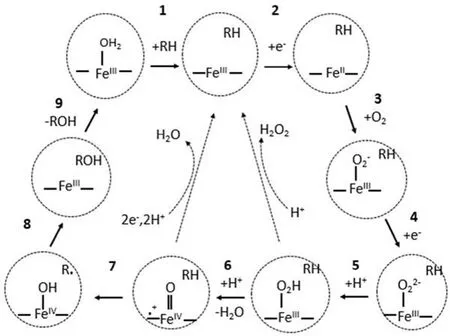
图1 CYP450 的催化循环
1.2 CYP450 BM3
CYP450 BM3 是由细胞色素单加氧酶和依赖 NADPH的 FMN/FAD 还原酶组成的单链融合蛋白。它的分子量大约为 119 kD,其中含有两分子黄素的多肽结构域为 65 kD,血红素结构域为 55 kD(图2)[15]。

图2 BM3 的结构(Heme 为 BM3 的血红素结构域;FMN 和 FAD 为含两分子黄素的还原酶结构域;数字代表氨基酸位点)
CYP450 BM3 能够将链长为 C12~C20的饱和脂肪酸的 ω-1、ω-2、ω-3 位羟基化,同时也能催化链长为 C12~C20的单不饱和脂肪酸、脂肪醇和脂肪酰胺等,但催化活力不高。此外,BM3 在大肠杆菌中高表达,作为一种血红素依赖的催化剂参与多种生理过程,如参与药物和外源性化学物质的代谢。系统进化分析表明,P450 BM3(CYP102)与人类主要参与药物代谢的酶,特别是 P450 3A4[16]具有较高的同源性。
目前,对于 P450 BM3 的研究主要集中在以下几个方面:①研究催化反应机制与酶结构的关系;②通过蛋白质工程增加底物的特异性,提高对底物的催化性能(KM、Kcat和偶联效率)及扩大底物作用范围;③寻找廉价的辅因子和电子转移系统;④开发高效、精确的高通量筛选方法。
虽然 P450 BM3 是已知的最活跃的 CYP450,但事实上非天然底物往往具有低的 NADPH 消耗率和不良的偶联率[17],这种酶仍然不能像野生型的酶催化它们优选的脂肪酸底物那样有效地催化甾体,野生型酶的效用受到限制。
2 催化甾体羟基化的 P450 氧化酶 BM3 的蛋白质工程
定向进化和(饱和)定点突变是蛋白质工程的主要研究手段,用以提高酶催化的精确性和有效性,弥补天然酶活性低、稳定性差及缺乏商业利用价值的缺陷。蛋白质工程可以帮助人们对目标蛋白进行有目的的改造,因此许多研究希望通过蛋白质工程获得 P450 BM3 羟基化甾醇的催化性能,并且借助新突变酶结构和功能的关系为酶的进一步研究改造提供更多有价值的信息。过去几年通过蛋白质工程已成功得到许多 P450 BM3 的突变体,特别是那些对一系列甾体底物有影响的突变体,活性增强的突变让这种自给自足的系统的催化潜力得到更充分利用。P450 BM3 的蛋白质工程在甾体羟基化的改良领域中所作的尝试以及获得的成果为BM3 羟基化疏水性甾体的深入研究提供了理论依据。
2.1 催化甾体羟基化的 P450 氧化酶 BM3 的定向进化
目前在大量蛋白质结构和功能信息严重缺乏的情况下,酶分子的体外定向进化技术不需要事先了解蛋白质空间结构和功能之间的关系,适合一切蛋白质的改造,极大拓宽了蛋白质工程的研究和应用范围。过去 20年中,定向进化实验表明单个氨基酸的替换通常可以提高酶的催化活性或稳定性,并且可通过有益突变的积累使酶分子获得显著的活性变化(改变 1%~2% 的序列)[8]。
BM3 定向进化的基本原理即在选定的 BM3 或其突变体的基础上,根据需要进行基因的突变或重组,使基因发生变异,再通过一定筛选方法获得预先期望的具有某些特性(如甾体特定位置的羟基化产物)的进化酶。
在 BM3 定向进化的策略中,两个最关键的环节是产生分子多样性和定向筛选,即突变体文库的构建和目标表型筛选。
2.1.1 突变体文库的构建 为了得到多样性分子结构,建立突变体文库最广泛采用的方法是随机突变的易错 PCR[18]。例如,以 BM3 的三突变体 R47L/F87V/L188Q 为模板,经过第一轮易错 PCR 筛选得到突变体 M01、M02,以 M01为模板经过两轮易错 PCR 筛选得到的 M11[19]能羟基化睾酮、黄体酮、去甲雄烯二酮等甾体[20-21],并且形成不同构型和不同位置的羟基化产物。基于 1393(BM3 的突变体)随机突变得到的突变体 5B10 能使黄体酮 16,17 位环氧化。16,17-环氧化是甾体激素合成的重要中间体[22]。环氧化疏水性甾体在制药行业甾体的合成中发挥重要作用。
2.1.2 突变体文库的筛选 在突变体文库产生之后,筛选的条件决定着蛋白质预期特征的进化方向。实际上,一旦多样性文库构建好后,定向进化实验成败的关键则在于是否有一个针对目标蛋白特定功能的高效筛选方法。
文献报道了一些关于 CYP450 的活性筛选方法。这些方法包括耗时的和昂贵的色谱光谱和放射化学技术[23-25],但这些方法只适用纯酶的筛选[26],因此不适合进行高通量筛选。而在一些报道的高通量筛选方法中[27-28],筛选系统依赖酶对特定底物的转化率,因此应用范围窄。
在定向进化中最常用的筛选仪器是高通量微孔板酶标仪。根据底物或产物的反应特征,高通量 96 微孔板可快速、自动、定量地鉴定酶的底物特异性、热稳定性等性状[29]。Tsotsou 等[30]报道一种新的用微孔板筛选 P450 BM3 突变体的方法,称为碱法筛选。此法可用于所有同 NADPH 相偶联的酶,也可对全细胞(即未破碎的细胞)催化进行检测,并且对各种类型的 P450 以及各种潜在的底物均可进行筛选。
但对于那些难以或无法建立筛选模型的蛋白质来说,定向进化的策略难以奏效。许多 P450 酶参与没有或很少有官能基团烃的氧化,如链烷烃、萜类、甾体或脂肪酸。因此,敏感而高效的筛选方法成为体外定向进化蛋白质的瓶颈,有必要继续开发新的高通量、自动化、定量表征酶活力的筛选方法。
2.2 催化甾体羟基化的 P450 氧化酶 BM3 的(饱和)定点突变
2.2.1 定点突变 定点突变是酶基因修饰的重要方法,也是研究蛋白质结构与功能的一项非常有价值的工具。定点突变首先需要合成特定的引物,该引物一端含有需要引入的突变,另一端含有同目的 DNA 互补的同源序列,而后用 PCR的方法在 DNA 聚合酶的作用下通过引物同模板发生同源重组而引入需要的突变,最后筛选得到目的突变体。由于定点突变是在清楚了解酶结构、功能、催化机制的基础上进行的,能在预先设计的位置建立起突变基因文库,缩小了有效突变基因文库的大小,减少工作量,但通常需预先通过理性设计预测或用易错 PCR 技术筛选到潜在的重要氨基酸残基位点。
定点突变包括单点突变和组合突变。将 P450 BM3 的2 个突变体(M01 和 M11)的 82 位丙氨酸突变为色氨酸后,M01A82W(M11A82W)均提高了对睾酮和炔诺酮 16β羟基化的选择性[20]。而将 M01A82W 突变体的 72 位丝氨酸突变为异亮氨酸后,产物的立体选择性改变,可以制备高产率 16α-羟基睾酮(产率达到 81%)[31]。此外,Kille 等[32]通过组合文库 A、B 和 C 中活性位点的突变获得甾醇的具有位置选择性和立体选择性的羟基化产物。
2.2.2 饱和定点突变 饱和定点突变是一种快速特异的蛋白质工程改造方法,通过把蛋白质的每一个氨基酸都替换为其他 19 种天然氨基酸来实现。饱和突变通常是在“热点”氨基酸上进行操作。在许多突变体中,单个氨基酸残基位点的改变即可提高酶的热稳定性或催化效率。将 CYP102A1突变体 M11 的 87 位苯丙氨酸突变为其他 19 种氨基酸时,发现不同的突变体能在不同的部位羟基化睾酮,且不同产物的得率和活性有较大差异[33]。
2.3 催化甾体羟基化的 P450 氧化酶 BM3 的蛋白质工程的应用
Venkataraman 等[21]通过使用红球菌突变株 RG9 表达P450 BM3 突变体 M02[19]来评估全细胞生物转化去甲雄烯二酮(NAND)。经 NMR 证实,纯化的 P450 BM3 突变体M02 不但以 95% 的区域选择性催化 NAND 形成 16β羟基化去甲雄烯二酮,而且可以得到 16β 产物。全细胞转化 1 g/L 的 NAND 可以生产出 0.35 g/L 16β 羟基化去甲雄烯二酮。全细胞红球菌突变株 RG9 本身不转化 NAND,表明代谢产物是由 P450 BM3 突变体 M02 催化。研究表明红球菌是一种新颖和高效的异源表达具有高度选择性和羟基化甾体活性的 BM3 M02 酶的宿主。
3 结语
目前蛋白质工程应用的领域逐渐扩大,涉及基因治疗、医学诊断、食品和化学工业等各方面,正在产生巨大的商业利益。而且,各种新型突变技术和新型检测系统的出现,推动着蛋白质工程研究向纵深发展。
本文探讨了催化甾体羟基化的 P450 氧化酶 BM3 蛋白质工程的研究进展。通过新一代测序技术和生物信息学分析等方法,筛选出参与甾体羟基化的相关 CYP450,并对候选基因进行功能验证,进一步阐明甾体羟基化的可行性和重要性,使羟基化甾体的制备变得越来越容易,特别是满足制药行业对光学活性化合物日益增长的需求。利用蛋白质工程获得的 BM3 突变体通过使用单一的酶使甾体羟化有了区域选择性和立体选择性,从而提供甾体羟基化这一珍贵转化的方便平台。
但是,由于甾体底物生物溶解性差,底物产物的反馈抑制和全细胞生物催化副产物多等原因,甾体的羟基化效率仍然偏低,并且许多工作仍需要完善以提高潜在的生物催化剂的催化性能。P450 氧化酶 BM3 的一些活性位点的作用机制尚未完全阐明,突变体以何种方式影响底物结合,酶的活性和选择性并不总是显而易见。此外,BM3 的催化性能高度依赖含有两个黄素辅因子的融合还原酶介导的 NADPH到甾体底物的电子转移效率[34]。用来催化电子传递的NADPH 辅因子也显著影响扩大生物催化过程的成本,所以用于药物代谢物的 CYP450 氧化酶 BM3 还没有达到工业生产的要求。
[1] Tong WY, Dong X. Microbial biotransformation: recent developments on steroid drugs. Recent Pat Biotechnol, 2009, 3(2):141-153.
[2] Cheng HX, Zhang GL. Progress in research of the interaction between the steroid hormone drugs and cytochrom P450 metabolic enzymes. Chin J Clin Pharmacol, 2015, 31(13):1328-1330. (in Chinese)
程海旭, 章国良. 甾体激素类药物与细胞色素P450代谢酶系相互作用的研究进展. 中国临床药理学杂志, 2015, 31(13):1328-1330.
[3] Donova MV, Egorova OV. Microbial steroid transformations: current state and prospects. Appl Microbiol Biotechnol, 2012, 94(6):1423-1447.
[4] Ortiz de Montellano PR. Cytochrome P450: structure, mechanism,and biochemistry. New York: Kluwer Academic/Plenum Publishers,2005.
[5] Urlacher VB, Girhard M. Cytochrome P450 monooxygenases: anupdate on perspectives for synthetic application. Trends Biotechnol,2012, 30(1):26-36.
[6] Torres Pazmiño DE, Winkler M, Glieder A, et al. Monooxygenases as biocatalysts: Classification, mechanistic aspects and biotechnological applications. J Biotechnol, 2010, 146(1-2):9-24.
[7] van Beilen JB, Funhoff EG. Expanding the alkane oxygenase toolbox:new enzymes and applications. Curr Opin Biotechnol, 2005, 16(3):308-314.
[8] Lan B, Kang LL. Overview of the genetic polymorphism of cytochrome P450 2C19. Foreign Med Sci (Sect Medgeography), 2015,36(2):159-164. (in Chinese)
兰冰, 康龙丽. 细胞色素P450 2C19遗传多态性的研究进展. 国外医学医学地理分册, 2015, 36(2):159-164.
[9] Arnold FH. How proteins adapt: lessons from directed evolution. Cold Spring Harb Symp Quant Biol, 2009, 74:41-46.
[10] Miles CS, Ost TW, Noble MA, et al. Protein engineering of cytochromes P-450. Biochim Biophys Acta, 2000, 1543(2):383-407.
[11] Behrendorff JB, Huang W, Gillam EM. Directed evolution of cytochrome P450 enzymes for biocatalysis: exploiting the catalytic versatility of enzymes with relaxed substrate specificity. Biochem J,2015, 467(1):1-15.
[12] Eiben S, Kaysser L, Maurer S, et al. Preparative use of isolated CYP102 monooxygenases -- a critical appraisal. J Biotechnol, 2006,124(4):662-669.
[13] Mowat CG, Gazur B, Campbell LP, et al. Flavin-containing heme enzymes. Arch Biochem Biophys, 2010, 493(1):37-52.
[14] Whitehouse CJ, Bell SG, Wong LL. P450(BM3) (CYP102A1):connecting the dots. Chem Soc Rev, 2012, 41(3):1218-1260.
[15] Fasan R, Chen MM, Crook NC, et al. Engineered alkane-hydroxylating cytochrome P450(BM3) exhibiting nativelike catalytic properties. Angew Chem Int Ed Engl, 2007, 46(44):8414-8418.
[16] Di Nardo G, Gilardi G. Optimization of the bacterial cytochrome P450 BM3 system for the production of human drug metabolites. Int J Mol Sci, 2012, 13(12):15901-15924.
[17] Whitehouse CJ, Bell SG, Yang W, et al. A highly active single-mutation variant of P450BM3 (CYP102A1). Chembiochem,2009, 10(10):1654-1656.
[18] Leung DW, Chen E, Goeddel DV. A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction. Technique, 1989, 1:11-15.
[19] van Vugt-Lussenburg BM, Stjernschantz E, Lastdrager J, et al. Identification of critical residues in novel drug metabolizing mutants of cytochrome P450 BM3 using random mutagenesis. J Med Chem,2007, 50(3):455-461.
[20] Rea V, Kolkman AJ, Vottero E, et al. Active site substitution A82W improves the regioselectivity of steroid hydroxylation by cytochrome P450 BM3 mutants as rationalized by spin relaxation nuclear magnetic resonance studies. Biochemistry, 2012, 51(3):750-760.
[21] Venkataraman H, Te Poele EM, Rosloniec KZ, et al. Biosynthesis of a steroid metabolite by an engineered Rhodococcus erythropolis strain expressing a mutant cytochrome P450 BM3 enzyme. Appl Microbiol Biotechnol, 2015, 99(11):4713-4721.
[22] Jiang D, Tu R, Bai P, et al. Directed evolution of cytochrome P450 for sterol epoxidation. Biotechnol Lett, 2013, 35(10):1663-1668.
[23] Schwaneberg U, Otey C, Cirino PC, et al. Cost-effective whole-cell assay for laboratory evolution of hydroxylases in Escherichia coli. J Biomol Screen, 2001, 6(2):111-117.
[24] Neufeld K, Zu Berstenhorst SM, Pietruszka J. Evaluation of coumarin-based fluorogenic P450 BM3 substrates and prospects for competitive inhibition screenings. Anal Biochem, 2014, 456:70-81.
[25] Moriya T, Kawamata A, Takahashi Y, et al. An improved fluorogenic NAD(P)+ detection method using 2-acetylbenzofuran: its origin and application. Chem Commun (Camb), 2013, 49(98):11500-11502.
[26] Maves SA, Yeom H, McLean MA, et al. Decreased substrate affinity upon alteration of the substrate-docking region in cytochrome P450(BM-3). FEBS Lett, 1997, 414(2):213-218.
[27] Schwaneberg U, Schmidt-Dannert C, Schmitt J, et al. A continuous spectrophotometric assay for P450 BM-3, a fatty acid hydroxylating enzyme, and its mutant F87A. Anal Biochem, 1999, 269(2):359-366.
[28] Alcalde M, Farinas ET, Arnold FH. Colorimetric high-throughput assay for alkene epoxidation catalyzed by cytochrome P450 BM-3 variant 139-3. J Biomol Screen, 2004, 9(2):141-146.
[29] Glieder A, Meinhold P. High-throughput screens based on NAD(P)H depletion. Methods Mol Biol, 2003, 230:157-170.
[30] Tsotsou GE, Cass AE, Gilardi G. High throughput assay for cytochrome P450 BM3 for screening libraries of substrates and combinatorial mutants. Biosens Bioelectron, 2002, 17(1-2):119-131.
[31] Venkataraman H, Beer SB, Bergen LA, et al. A single active site mutation inverts stereoselectivity of 16-hydroxylation of testosterone catalyzed by engineered cytochrome P450 BM3. Chembiochem, 2012,13(4):520-523.
[32] Kille S, Zilly FE, Acevedo JP, et al. Regio- and stereoselectivity of P450-catalysed hydroxylation of steroids controlled by laboratory evolution. Nat Chem, 2011, 3(9):738-743.
[33] Vottero E, Rea V, Lastdrager J, et al. Role of residue 87 in substrate selectivity and regioselectivity of drug-metabolizing cytochrome P450 CYP102A1 M11. J Biol Inorg Chem, 2011, 16(6):899-912.
[34] Bloom JD, Labthavikul ST, Otey CR, et al. Protein stability promotes evolvability. Proc Natl Acad Sci U S A, 2006, 103(15):5869-5874.
10.3969/j.issn.1673-713X.2015.06.014
天然药物活性物质与功能国家重点实验室自主课题重点项目(GTZA201404)
100050 北京,中国医学科学院北京协和医学院药物研究所天然药物活性物质与功能国家重点实验室/卫生部天然药物生物合成重点实验室
孔建强,Email:jiangqiangk@imm.ac.cn
2015-07-01
