气相色谱法测定比卡鲁胺中残留溶剂二甲基甲酰胺
2015-11-20阮昊陈悦
阮昊 陈悦
(浙江省食品药品检验研究院,浙江 杭州310004)
比卡鲁胺(Bicalutamide)是英国阿斯利康公司研制的一种新型口服非甾体类雄性激素受体阻断剂,主要用于治疗晚期前列腺癌[1]。 比卡鲁胺国内现有生产企业3 家,本次研究收集了国内3 个生产厂家的合成工艺,共涉及8 种溶剂。 其中,第2类溶剂4 种:二氯甲烷、正己烷、四氢呋喃和N,N-二甲基甲酰胺(DMF);第3 类溶剂4 种:乙醇、乙醚、丙酮和乙酸乙酯[2]。 为确保临床用药安全,应对上述残留溶剂进行控制。 因各厂家生产工艺不完全相同,因此所需控制的残留溶剂也有所不同,因DMF 沸点较其余7 种溶剂高,需用直接进样法测定,因此为提高测定方法的适用性,本研究建立了可测定DMF 残留溶剂的直接进样气相色谱分析方法。 现报道如下。
1 仪器与试药
配备氢火焰离子化检测器(FID)的Agilent 7890A 气相色谱仪;Mettler AE163 型电子分析天平。
DMF,N-甲基吡咯烷酮均为色谱纯。
2 方法与结果
2.1 色谱条件
色谱柱:INNOWAX 型(涂布聚乙二醇固定液)弹性石英毛细管柱(30.0 m × 0.25 mm,0.25 μm),柱温:程序升温模式,初始60 ℃,以10 ℃/min 的速率升温至200 ℃,保持3 min;FID检测器,检测器温度为250 ℃;分流进样,分流比为20∶1,进样口温度为200 ℃;载气为氮气,流速为1.0 mL/min;直接进样,进样体积为1.0 μL。
2.2 溶液的制备
2.2.1 混合对照品溶液的制备 称取DMF 226.40 mg,置25 mL 量瓶中,用N-甲基吡咯烷酮溶解并定容,作为对照品储备液。精密量取浓储备液1 mL,置100 mL 量瓶中,加N-甲基吡咯烷酮稀释至刻度,摇匀,即成每1 mL 中含DMF 约88 μg 的对照品溶液。
2.2.2 供试品溶液的制备 取本品约0.5 g,精密称定,置5 mL 量瓶中,加N-甲基吡咯烷酮溶解并稀释至刻度,即得。
2.2.3 空白溶液 取空白溶剂N-甲基吡咯烷酮作为空白溶液。
2.3 系统适用性试验
因DMF 在生产过程中会伴随N,N-二甲基乙酰胺(DMA)带入,故考察DMF 和DMA 的分离度作为系统适用性试验。 称取23.10 mg DMF 和26.13 mg DMA 置25 mL 量瓶中,用N-甲基吡咯烷酮溶解并定容,精密量取该溶液2 mL,置20 mL量瓶中,加N-甲基吡咯烷酮稀释至刻度,摇匀,即成每1 mL 中含DMF 88 μg 和DMA 约88 μg 的混合溶液,作为系统适用性溶液,直接进样,记录色谱图,结果色谱图中DMF 和DMA 的分离度为8.56;其理论塔板数分别为72 419 和57 319。 空白溶液在与DMF 相同保留时间的位置无色谱峰出现,溶剂对本品残留溶剂的测定无干扰。 见图1。
2.4 线性关系考察
精密量取“2.2.1”项下对照品储备液适量,加N-甲基吡咯烷酮分别稀释1 系列6 个浓度的对照品溶液,按“2.1”项下的色谱条件进样测定。 以对照品浓度为横坐标X,被测溶剂峰面积为纵坐标Y,进行线性回归分析,得回归方程为:
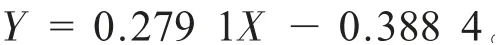
DMF 在22.64 ~226.40 μg/mL 范 围 内 峰 面积与浓度呈良好的线性关系,r= 0.999 8。
2.5 色谱系统的精密度
取“2.2.1”项下混合对照品溶液,按“2.1”项下的色谱条件进样测定6 次,量取各色谱图中DMF的峰面积,对色谱系统的精密度进行考察。结果测得DMF 的RSD 为0.8%(n= 6),表明系统的精密度明显优于《中国药典》对气相色谱顶空进样法系统适用性的最低要求。
2.6 重复性试验
取厂家1 提供的A140301 批次样品(DMF 本底为0),精密称取6 份,用“2.2.1”项下对照品溶液溶解并定量稀释制成每1 mL 中约含比卡鲁胺0.1 g 的溶液,按“2.1”项下的色谱条件进行测定,其结果分别为0.082 8%,0.081 9%,0.075 1%,0.077 9%,0.081 2%及0.083 4%,RSD 为3.9%(n=6)。
2.7 回收率试验
取厂家1 的A140301 批次样品(DMF 本底为0)进行试验。 取“2.2.1”项下制备的对照品溶液作为相当于限度100%的加样溶液Sm,精密量取该对照品溶液10 mL,用N-甲基吡咯烷酮稀释定容至20 mL,配制成相当于限度50%的加样溶液Sl;精密量取“2.2.1”项下制备的对照品储备液3 mL,用N-甲基吡咯烷酮稀释定容至200 mL,配制成相当于限度150%的加样溶液Sh。 精密称取厂家1 提供的A140301 批次样品9 份,各约0.50 g,置量瓶中,每个浓度各3 份,分别用上述3 个浓度的加样溶液溶解并定容,即得平行制备的一组共9份回收率试验用加样测定溶液。取上述9 份试样,按“2.1”项下的色谱条件进样测定,量取各色谱图中DMF 的峰面积,以标准曲线法计算。 分别扣除供试品原液中DMF 的本底残留后,计算出DMF 的回收率。 结果测得DMF 的平均回收率为91.9%,RSD 为2.7%,平均回收率均在80% ~120%之内,符合残留溶剂测定准确度的要求。试验结果表明,本法用于DMF 的残留量测定具有较好的准确性。
2.8 灵敏度试验
精密量取对照品储备液适量,用N-甲基吡咯烷酮逐步稀释并按“2.1”项下的色谱条件进样测定,结果测得DMF 的检测限(S/N = 3)为5.9 μg/mL,定量限(S/N = 10)为22.6 μg/mL。
2.9 样品测定
取3 个厂家共8 批产品,按“2.2”项下的方法制备混合对照品溶液和供试品溶液,按“2.1”项下的色谱条件进样分析,隔数日后再次同法测定。按外标法以峰面积计算DMF 的残留量。测定结果见表1。 各批供试品的测定结果均低于《中国药典》2010 年版二部附录ⅧP 规定的限度[3],即DMF应≤0.088%。
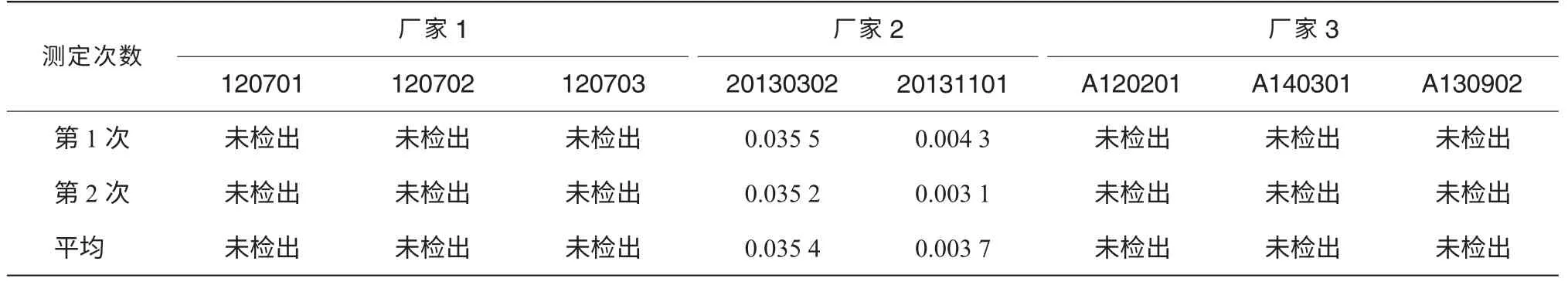
表1 比卡鲁胺样品中残留溶剂DMF 测定结果 (%)
3 讨论
3.1 色谱柱的选择
试验了6%氰丙基苯基-94%二甲基聚硅氧烷(DB-624)柱,结果峰形很差;用聚乙二醇固定液柱,DMF 峰形较好,保留时间合适,可与DMA完全分离。 因此色谱柱选择聚乙二醇固定液(INNOWAX)柱。
3.2 柱温的选择
①柱温为120 ℃时,DMF 峰与其他峰共出,无法分离;②故降低柱温,60 ℃保持6 min 后,以15 ℃/min 升温至200 ℃,DMF 保留时间为14 min,出峰较迟,基线噪音较大;③因条件②出峰较晚,故除去保持时间,直接以60 ℃起始,以15 ℃/min升温至200 ℃,DMF 5 min 出峰,保留时间合适,但基线噪音仍较大;④因基线噪音大,故降低升温速率,以60 ℃起始,以10 ℃/min 升温至200 ℃,DMF 6 min 出峰,保留时间合适,基线噪音较小。故选择条件④作为程序升温条件,DMF 可与相邻峰完全分离,与类似物DMA 也可良好分离。
3.3 溶解介质的选择
比卡鲁胺与DMF 水溶性均差,不能直接用水作溶剂,且DMF 在水中峰形较差,而在含N 溶剂,如N-甲基吡咯烷酮中,则峰形较好。 经试验,用N-甲基吡咯烷酮为溶剂,可将比卡鲁胺和被测溶剂完全溶解,峰形较好,基质效应小,回收率好,因此选择N-甲基吡咯烷酮作为溶剂。
[1] Tucker H,Crook JW,Chesterson GJ. Nonsteroidal antiandrogens. Synthesis and structure-activity relationships of 3-substituted derivatives of 2-hydroxypropionanilides[J]. J med Chem,1988,31(5):954-959.
[2] ICH 指导委员会. 药品注册的国际技术要求[M]. 北京:人民卫生出版社,2007:134-136.
[3] 国家药典委员会.中华人民共和国药典(二部)[M]. 北京:中国医药科技出版社,2010:附录61-65.
