ClC-0氯离子通道蛋白质空间结构的同源建模
2015-11-19朱紫洪郝栋梁
于 涛,朱紫洪,郝栋梁,彭 黎,张 攀,徐 号,郭 旭
(江汉大学 物理与信息工程学院,湖北 武汉 430056)
ClC-0氯离子通道蛋白质空间结构的同源建模
于 涛,朱紫洪,郝栋梁,彭 黎,张 攀,徐 号,郭 旭
(江汉大学 物理与信息工程学院,湖北 武汉 430056)
ClC型氯离子通道蛋白质在调节生命体的新陈代谢过程中起着重要作用。在原有蛋白质ClC-ec1晶体结构的基础上,依据同源建模的原理,借助SWISS-MODEL网络服务器构建了一类重要氯离子通道蛋白质ClC-0的空间结构,同时对所得结构的合理性进行了分析。通过衡量结构的Z-score值、分析结构的空间坐标波动性以及利用视图软件VMD将建模的ClC-0结构和模板结构进行比较,发现通过同源建模所得到蛋白质ClC-0的空间结构符合后续研究的要求,这在很大程度上方便了该类蛋白质的后续计算模拟研究。
同源建模;氯离子通道蛋白质;SWISS-MODEL
0 引言
离子通道是镶嵌在生物体细胞膜上一类重要的蛋白质微小孔道,它在生命体系的新陈代谢过程中扮演着极其重要的角色。氯离子通道(chloride ion channel,CIC)是其中极为重要的一类阴离子通道,它担负着包括盐类离子的跨膜输运、调节细胞pH值、控制细胞体积、调节电脉冲以及可激肌肉细胞的电压稳定性等重要的生理和细胞功能[1]。研究者们通过生化、物理数学以及计算机模拟计算等技术方法对ClC类氯离子通道进行了广泛的研究,这些研究包括结构分析、功能表达、变异导致相关疾病等。研究氯离子通道的结构与功能关系已成为保护人类健康,揭示致病机制和开发新药的前沿热点课题。
在ClC型氯通道蛋白质家族中,有两类蛋白质最具有代表性,同时也是被研究最广泛的两类氯通道蛋白质,它们分别是大肠埃希菌(Escherichia coli)氯离子通道蛋白质ClC-ec1和电鳐鱼类电器官氯通道蛋白质ClC-0[2-3]。对于蛋白质ClC-ec1,目前普遍认为是一类Cl-/H+相互输运的输运体(transporter),而ClC-0则被认为是一类典型的氯离子通道[4]。一直以来,研究者们认为ClC-0的门控(通道打开和关闭的过程)与目标离子的传导过程密切相关。ClC-0的注入和门控之间的密切联系说明:ClC-0门控是个非平衡过程,它必须利用能量趋近于平衡,这个能量可以来源于跨膜的离子或电压梯度。ClC-0的电压相关性不是来源于通道蛋白质中带电残基的移动,而是来源于氯离子自身的跨膜移动[1]。许多实验结果表明,门控的激活过程与注入过程是平行进行的,虽然有关ClC-0的单通道微电流记录清楚地显示它是一个离子通道,但是有的研究显示ClC-0结构中也可能存在质子的输运[5]。转运体中离子的输运也是强烈依赖溶液中离子的浓度、紧密的耦合门控和离子注入,并且过程远离平衡态。
对于氯离子通道蛋白质ClC-0,由于其结构在结晶时非常不稳定而导致不能通过实验上的X射线衍射方法得到其结构信息,这对理论研究产生了很大的阻碍。2002年,DUTZLER等[6]用X射线衍射方法测得了大肠埃希菌氯离子通道ClC-ec1的晶体结构。氯离子通道成为继水通道、钾离子通道之后又一种拥有原子水平上结构数据的离子通道,它使得通过理论计算方法研究氯通道中离子的输运机制成为可能。研究者们虽然对与ClC-ec1属于同一蛋白质家族的ClC-0研究较多,但由于ClC-0实验上的晶体结构还没有被测得,使得其在理论模拟以及计算方面的研究相对实验研究落后很多。
1 蛋白质结构的同源建模
在预测蛋白质三维空间结构方面,同源建模方法取得了很大成功,应用最为广泛,因此蛋白质同源建模已经成为蛋白质结构分析的重要方法。一般同源性超过30%的蛋白质序列都可以建立相当精确的结构模型,序列的同源性越高则建立模型结构的准确性就越高[7-9]。一般同源建模法推测蛋白质结构需要以下6个步骤:①搜索模板蛋白;②目标蛋白与模板蛋白的序列比较;③主链结构建模;④环区建模;⑤侧链建模及优化;⑥整体结构优化及评估。这些步骤可以用不同的程序完成,也可以使用同源建模服务器或者自动建模程序模块完成。目前,互联网上提供了很多可利用同源建模技术预测蛋白质结构的服务器,例如SWISS-MODEL(Geneva)、3D-JIGSAW(London)、FAMS(Tokoy)和SDSCI(San Diego),其中SWISS-MODEL网络建模服务器使用最广泛[10-11]。
1.1 同源建模服务器SWISS-MODEL
SWISS-MODEL网络同源建模服务器是一个自动化建模并且对任何学术团体都免费的蛋白质结构模拟环境,用户可以登录网站进行蛋白质结构的同源建模,还可以用打分函数对已建模的蛋白质结构进行评估以确定其优劣。本文大部分工作都是在SWISS-MODEL服务器上实现,同时借助生物大分子可视化软件VMD对同源建模所构建的结构进行三维显示和比较[12]。SWISS-MODEL是一个有注解的蛋白质结构同源建模服务器,并且与专业蛋白质分析系统(Expert Protein Analysis System,ExPASy)网站紧密联系,目前数据库中约有30 000个蛋白质三维结构模型,该服务器已向全世界的生物化学和分子生物学学者提供蛋白质三维结构自动建模服务。在SWISS-MODEL服务器的Web界面上可供选择的同源建模方式有3种:Automated Mode、Alignment Mode以及Project Mode,用户可以根据不同的需要选择相应的建模方式。
1.2 ClC-0蛋白质结构同源建模任务提交
使用SWISS-MODEL服务器对ClC-0蛋白质进行三维结构建模时,先要在蛋白质序列数据库中搜索相似性足够高的同源序列,以便用来建立最初的结构模型。在蛋白质数据库中,笔者找到了和ClC-0属于同一蛋白质家族的ClC-ec1蛋白质,两者序列之间的相似度极高,并且ClC-ec1的三维晶体结构已经被精确测量。因此,ClC-ec1为笔者提供了可靠的模板。采用蛋白质序列比对软件Clustal-W对ClC-0和ClC-ec1氨基酸序列进行序列比对[4],通过添加空位和人工微调,得到了最佳的序列比对模式,最终使得两条序列相似度达到最高,其中ClC-0和ClC-ec1序列相同的氨基酸残基占25%,侧链性质相同的氨基酸占58%,这说明属于同一蛋白质家族的ClC-0和ClC-ec1同源性非常高。在完成序列比对后,就可以在SWISS-MODEL服务器上提交任务。由于笔者提前已将需要建模的序列和模板序列进行了比对,因此可以选择建模结果准确的比对模式(Alignment Mode)进行同源建模,在该模式中需要人工将已经比对好的序列提交到服务器。点击Alignment Mode按键之后,就会进入比对建模模式界面(见图1)。在相应的对话框里填入提交者的邮箱、工作任务名字以及已经序列比对得分最高ClC-0和ClC-ec1的序列。在这里蛋白质的序列是以FASTA格式输入的,最后点击“submit alignment”按钮提交初始建模任务。
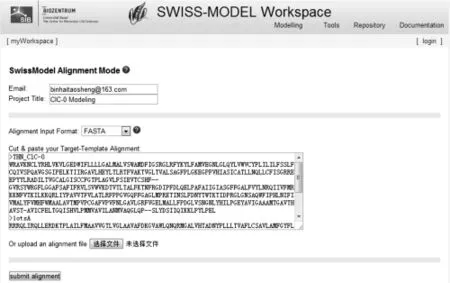
图1 在SWISS-MODEL服务器上提交建模任务的界面Fig.1 Interface of the submissin of modeling project on SWISS-MODEL web site
在完成任务的提交后就可以得到SWISS-MODEL服务器返回的序列比对结果(见图2)。在返回的比对结果中相同的氨基酸下面用星号“*”突出表示。图2是SWISS-MODEL服务器进行下一步同源建模的序列之间比对的最后结果,如果提交者对这一比对结果不满意,可以返回任务提交界面对序列比对进行调整,然后再提交。如果对序列比对的结果满意,就可以点击“submit alignment”按钮进行下一步建模。
2 ClC-0空间结构建模结果及分析
在提交任务后的0.5 h以内,任务提交者就会在邮箱里收到建模任务已完成的邮件。同时SWISSMODEL服务器的网页也发生了改变,显示出最终的建模结果以及对建模后结构的分析。SWISS-MODEL服务器对ClC-0蛋白质的结构建模结果如图3所示。在图3中显示的只是ClC-0蛋白质其中的A链的结构,该结构是根据序列比对结果进行建模,由于A链和B链氨基酸的序列完全相同,所以用相同的方法可以获得B链的同源建模结构。
为了说明建模结果的合理性,笔者对建模的结构进行了结构特性分析,通过在SWISS-MODEL服务器网页上查看和下载建模后的结构特征图,分析了ClC-0蛋白质整体建模结构的空间结构得分结果(Z-score,见图4),两个结构的Z-score值反映了建模结构和模板结构整体结构在能量上的差异性,通过图4可以看出,4类不同能量差异的Z-score值在-5左右,其中C-β相互作用能量差异为-5.11,所有原子相互作用能量差异为-5.43,溶剂化能量差异为-4.79,扭转角能量差异为-6.45。由于ClC-0蛋白质属于膜蛋白,细胞膜对蛋白质的空间约束较强,使得在没有细胞膜的约束下建模后结构的Z-Score值较低,对于膜蛋白来说这些值是合理的。这说明没有细胞膜的限制,ClC-0蛋白质的结构会出现局部变化,如果将建模后的结构放到细胞膜中,会使得整个结构的变化量大大减少,并且可以通过后续的分子动力学模拟将建模后的蛋白质和细胞膜实现完全融合。为了更详细地展示建模后结构的具体细节变化,笔者计算了建模后ClC-0蛋白质和模板蛋白质ClC-ec1之间随着氨基酸序列排序结构中原子空间坐标的差异性,图5是建模后ClC-0空间结构和模板结构在氨基酸序列上的每个氨基酸残基原子坐标的波动性变化情况。对于两端(C-端和N-端)处的氨基酸来说,由于跟其他氨基酸的相互作用偏弱,导致其结构的波动性偏大,处于蛋白质内部的疏水氨基酸的位置波动性大大减小,大部分氨基酸残基上所有原子的坐标偏差之和都小于5 Å。这说明通过同源建模得到的ClC-0蛋白质的结构基本跟模板ClC-ec1的结构相吻合。

图2 蛋白质ClC-0和ClC-ec1氨基酸序列比对结果Fig.2 Comparison of amino acid sequence of protein CIC-0 andClC-ec1

图3 SWISS-MODEL服务器对CIC-0建模结果的结构信息Fig.3 Structure information of ClC-0 protein by SWISS-MODEL server

图4 SWISS-MODEL服务器建模结果能量差异性的Z-score值Fig.4 The energy Z-score value of the modeling structure by SWISS-MODEL server

图5 SWISS-MODEL服务器建模结构和模板结构的原子坐标波动性分析Fig.5 Coordinates fluctuation analysis of the modeling structure and template structure
通过下载SWISS-MODEL服务器上ClC-0的建模结果中的原子坐标文件,用视图软件VMD做出了建模后的ClC-0结构示意图,通过跟模板ClC-ec1的结构进行比较,可以看出大部分结构都是一致的,只是在结构中转角处有部分差异,具体结果如图6所示(其中灰色区域为模板结构,螺旋结构为建模结构),这说明笔者事先通过序列比对,然后在SWISS-MODEL服务器上进行蛋白质空间结构的同源建模方法是可行的。

图6 建模ClC-0结构与模板ClC-ec1结构的差异性Fig.6 Differences between the modeling ClC-0 structure and the ClC-ec1 structure
3 结语
本研究从ClC-0氯离子通道蛋白质氨基酸序列出发,在提前进行序列比对的基础上,通过同源建模的方法第一次得到了该类蛋白质的可靠空间结构,同时对所构建的结构模型合理性进行了评估与分析。所得到的结构为后续研究(如系统静电分析、动力学模拟)提供了可靠样本,也为同源建模在研究蛋白质结构预测领域的应用奠定了基础。
(
)
[1]JENTSCH T J,NEAGOE I,SCHEEL O.ClC chloride channels and transporters[J].Current Opinion in Neurobiology,2005,15(3):319-325.
[2]陈丽娥,谢浩.ClC型氯离子通道的研究[J].生命的化学,2010,30(4):545-549.
[3]殷丽天,付月君,梁爱华.氯离子通道ClC家族的结构特点与功能研究[J].山西医科大学学报,2006,37(1):100-102.
[4]CORRY B,O′MARA M,CHUNG S H.Permeation dynamics of chloride ions in the ClC-0 and ClC-1 channels[J].Chemical Physics Letters,2004,386(4/5/6):233-238.
[5]LISAL J,MADUKE M.The ClC-0 chloride channel is a′broken′Cl-/H+antiporter[J].Nature Structural&Molecular Biology,2008,15:805-810.
[6]DUTZLER R,CAMPBELL E B,CADENE M,et al.X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity[J].Nature,2002,415(6869):287-294.
[7]曾炳佳,曹以诚,杜正平,等.同源建模关键步骤的研究动态[J].生物学杂志,2008,25(2):7-10.
[8]汤海旭,丁达夫.用于蛋白质同源模建及三维结构预测的结构比较方法[J].生物物理学报,1995,11(1):60-66.
[9]FISER A,DO R K G,SALI A.Modeling of loops in protein structures[J].Protein Science,2000,9(9):1753-1773.
[10]BIASINI M,BIENERT S,WATERHOUSE A,et al.SWISS-MODEL:modeling protein tertiary and quanternary structure using evolutionary information[J].Nucleic Acids Research,2014,12:252-258.
[11]谌容,陈敏,杨春贤,等.基于SWISS-MODEL的蛋白质三维结构建模[J].生命的化学,2006,26(1):54-56.
[12]HUMPHREY W,DALKE A,SCHULTEN K.VMD:visual molecular dynamics[J].Journal of Molecular Graphics,1996,14(1):33-38.
(责任编辑:胡燕梅)
Homology Modeling of Spatial Structure of CIC-0 Chloride Channel Proteins
YU Tao,ZHU Zihong,HAO Dongliang,PENG Li,ZHANG Pan,XU Hao,GUO Xu
(School of Physics and Information Engineering,Jianghan University,Wuhan 430056,Hubei,China)
CIC-type chloride ion channel proteins play important role in regulating the metabolism process of biological system.On the base of CIC-ec1 crystal structure,in accordance with the principle of homology modeling,using SWISS-MODEL net server,constructed the spatial structure of a kind of CIC-0 chloride ion channel protein,and analyzed the reasonableness of the obtained structure.Through measuring the Z-score of the structure,analyzing the fluctuation of space coordinates of the structure,and comparing the modeling CIC-0 structure with template structure,discovered that the spatial structure of CIC-0 protein by homology modeling complied with the requirement of further study,which makes the further simulation and computation be easy.
homology modeling;chloride channel protein;SWISS-MODEL
Q51;TP391.9
A
1673-0143(2015)02-0111-05
10.16389/j.cnki.cn42-1737/n.2015.02.003
2014-10-09
国家自然科学基金项目(11304123);国家留学基金项目(201408420100);江汉大学高层次人才科研启动经费项目(2013016);江汉大学学生科研项目(2013yb093)
于 涛(1982—),男,讲师,博士,研究方向:凝聚态物理、软物质物理。
