河西走廊石羊河下游地区盐碱土中放线菌多样性
——以民勤县为例
2015-11-19李海云牛世全孔维宝朱学泰张爱梅达文燕韩彩虹阎薇如西北师范大学生命科学学院甘肃兰州730070
李海云,牛世全,孔维宝,朱学泰,张爱梅,达文燕,韩彩虹,阎薇如,耿 晖(西北师范大学生命科学学院,甘肃 兰州 730070)
河西走廊石羊河下游地区盐碱土中放线菌多样性
——以民勤县为例
李海云,牛世全*,孔维宝,朱学泰,张爱梅,达文燕,韩彩虹,阎薇如,耿 晖(西北师范大学生命科学学院,甘肃 兰州 730070)
为了解甘肃省河西走廊石羊河流域地区盐碱土中放线菌种群结构及多样性,采用非培养法对河西走廊石羊河下游流域的3种不同类型土样(原生盐碱土、次生盐碱土和农田土)的总DNA进行提取,用放线菌特异性引物对16S rRNA基因进行扩增,构建放线菌16S rRNA克隆文库.用HaeⅢ和HhaⅠ两种限制性内切酶对阳性克隆子进行16S rDNA扩增片段限制性内切酶分析(Amplifed Ribosomal DNA Restriction Analysis,ARDRA),提取酶切带型不同的菌液进行测序,构建其系统发育树并进行多样性指数分析.结果显示:原生盐碱土克隆文库中90个阳性克隆分归于20个OTUs,分属于微球菌科(Micrococcaceae)、中村氏菌科(Nakamurellaceae)、类诺卡氏菌科(Nocardioidaceae)、链霉菌科(Streptomycetaceae)、棒状杆菌科(Corynebacteriaceae)、诺卡氏菌科(Nocardiaceae)和未知类群;次生盐碱土克隆文库中98个阳性克隆分归于32个OTUs,分属于纤维素单胞菌科(Cellulomonadaceae)、微球菌科(Micrococcaceae)、地嗜皮菌科(Geodermatophilaceae)、类诺卡氏菌科(Nocardioidaceae)、小单孢菌科(Micromonosporaceae)、伪诺卡氏菌科(Pseudonocardiaceae)、链霉菌科(Streptomycetaceae)、链孢囊菌科(Streptosporangiaceae)、高温单孢菌科(Thermomonosporaceae)、动孢囊菌科(Kineosporiaceae)、糖霉菌科(Glycomycetaceae)和未知类群;农田土克隆文库中98个阳性克隆分归于10个OTUs,分属于微球菌科(Micrococcaceae)、博戈里亚湖菌科(Bogoriellaceae)、地嗜皮菌科(Geodermatophilaceae)、中村氏菌科(Nakamurellaceae)、类诺卡氏菌科(Nocardioidaceae)和未知类群.其中,微球菌亚目(Micrococcineae)是3种不同类型土壤中的优势类群.多样性指数和稀释性曲线分析结果显示,3种不同类型土壤中放线菌多样性为次生盐碱土>原生盐碱土>农田土.
河西走廊;盐碱土;克隆文库;放线菌多样性
土壤微生物作为生态系统的重要组成部分,在有机物分解转化过程中占主导作用,具有巨大的生物化学活力,能动地影响生态系统中的能量流动和物质转化过程,并通过自身代谢对土壤结构和营养物质转化产生重要影响.而放线菌作为一类具有重大实用价值的微生物资源,广泛存在于不同的自然生态环境之中.在已知的微生物活性物质中,大约70%是由放线菌产生[1],在当前从常规放线菌寻找开发新化合物日益困难的情况下,不少学者已经把目光投向对极端环境放线菌的研究[2-5].这类放线菌能长期生长在高温、低温、高酸、高碱或高盐等极端特异环境中,具有独特的基因类型、特殊的生理机制,从而产生特殊的代谢产物[6],这也为开发利用极端环境条件下放线菌资源提供了物质资源.
河西走廊地区是我国西北地区的棉粮基地.由于该地区特殊的地质条件和不合理的耕灌制度,土体内盐水运动加快,加之蒸发量大,盐分聚表,土壤次生盐渍化现象十分普遍,造成粮食产量大幅度降低,甚至部分耕地不得不弃耕[7].对于河西走廊盐碱土中微生物多样性的研究,已有的报道主要是对细菌种群多样性的研究,如牛世全等[8]的盐碱土微生物功能群季节动态与土壤理化因子的关系,以及河西走廊春季不同盐碱土壤中微生物数量、酶活性与理化因子的关系[9],这些工作主要集中在功能性菌群和土壤理化因子的关系方面,而有关该地区土壤中放线菌种群多样性的分子生态学研究还尚未见报道.基于培养方法很难全面反映出环境中微生物的多样性信息,本研究通过未培养技术对河西走廊石羊河流域下游地区的原生盐碱土、次生盐碱土和农田土中放线菌多样性进行分析研究,能够更加真实地反映出盐碱土壤中放线菌的生态分布,以弥补河西走廊盐碱土中放线菌多样性研究的不足.
1 材料与方法
1.1 研究区域概况
民勤县(101°49′~104°12′E,38°03′~39°28′N),位于甘肃省河西走廊东北部,石羊河流域下游,南邻凉州区,西毗金昌,东、西、北3面与内蒙古自治区接壤.处于腾格里和巴丹吉林2大沙漠之间.全县总土地面积1.6×106hm2,其中沙漠、戈壁、剥蚀山地和盐碱滩地等占91%,绿洲仅占9%,绿洲边缘风沙线长达408km.气候属温带干旱气候区,多年平均降水量110mm,蒸发量为2650mm.民勤县境内唯一的地表水资源为石羊河,民勤绿洲是该县主要农业区,自然植被大致划分为荒漠和草甸2大类型,属较典型的极干旱沙漠荒漠草原植被区,生态环境十分脆弱[10-12].
1.2 材料
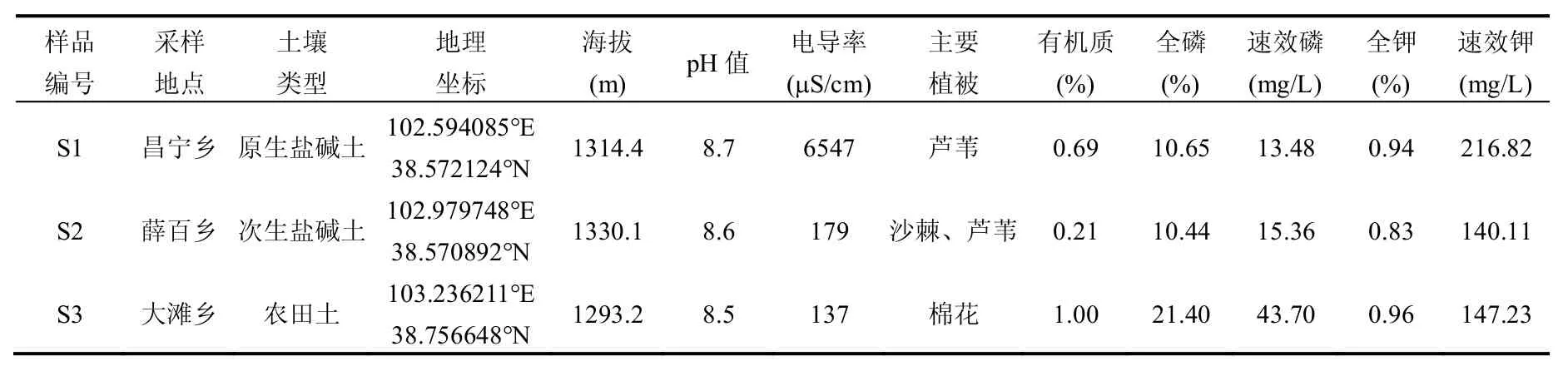
表1 试验所用土壤样品基本信息Table 1 Basic information of the tested soil samples
1.1.1 样品来源与采集 土壤样品采集于2012年12月甘肃省民勤县分布的盐碱土(表1),采用五点取样法采集土样,即去除地表土,取0~20cm的土壤样品,每个点取样量大体一致,土样混匀后收集,采集土样保存于灭菌的密封袋中,并记录采集人姓名、采集时间、地点、地上主要植被等信息,置于冰袋上冷藏迅速带回实验室,在-20℃冰箱中保存备用.经过试验分析测定,土壤样品基本信息见表1.
1.1.2 主要试剂与器材 DNA胶回收纯化试剂盒(北京全式金生物技术有限公司),Premix Ex Taq DNA聚合酶(大连 TaKaRa),pMD18-T载体(大连 TaKaRa),Hae Ⅲ和HhaⅠ限制性内切酶(大连 TaKaRa),PCR扩增仪(美国Bio-Bad),凝胶成像系统(美国Bio-Bad).
1.2 方法
1.2.1 总DNA的提取纯化及16S rRNA PCR扩增 盐碱土壤微生物总DNA的提取按照Zhou等[13]的方法并稍作改动进行提取.将提取的粗DNA样品,以λDNA/Hind Ⅲ为DNA Maker,用1%琼脂糖凝胶进行电泳检测,将DNA凝胶电泳图谱上的条带用凝胶回收试剂盒进行切胶回收.利用放线菌特异性引物[14](S-20:5′-CGCGG CCTATCAGCTTGTTG-3′,A-19:5′-CCGTACTCCCCAGGCGGGG-3′)对河西走廊盐碱土壤样品总DNA进行16S rRNA基因扩增,PCR反应条件参照Stach等[14]的方法.PCR扩增反应总体系为25µL:上下游引物各(10mmol/µL)1µL,模板 2µL,Premix Ex Taq DNA聚合酶 12.5µL,补加ddH2O至25µL.反应参数:94℃变性5min;94℃ 45s,58℃ 45s,72℃90s,35个循环,72℃延伸10min.PCR扩增产物在1%琼脂糖凝胶中进行电泳检测.并将目的片段用DNA凝胶回收试剂盒进行切胶回收纯化.
1.2.2 16S rRNA基因文库构建及阳性克隆筛选 经纯化后的PCR产物利用T4DNA连接酶与pMD18-T vector进行连接,转化到大肠杆菌DH5α感受态细胞中,转化产物涂布在含有氨苄青霉素/IPTG/X-Gal的LB平板上,在37℃条件下培养16~24h,随机挑取白色克隆子,并构建基因文库.用pMD-18T vector通用引物M13-47和M13-48对挑取的克隆子对应菌液进行PCR扩增检测,筛选的阳性克隆子菌液,保存在4℃条件下备用.
1.2.3 ARDRA分析与测序 用HaeⅢ、HhaⅠ限制性内切酶对阳性克隆子菌液PCR产物进行酶切,酶切体系均为20µL:10×Buffer 2µL;HaeⅢ或HhaⅠ 1µL;PCR扩增产物7µL;ddH2O 10µL,37℃酶切1h.酶切产物用1%的琼脂糖电泳检测.酶切图谱用Quantity One软件进行分析,分析产物经酶切后的谱型,计算每个谱型出现的频率,将具有不同谱型的克隆子进行测序.所测序列用Mothur进行嵌合体检验并去除嵌合体序列[15].
1.2.4 系统发育分析及序列登录号 将获得的序列与GenBank数据库进行比较,选取相似性最高的16S rRNA基因序列,运行ClustalX[16]进行多序列比对,根据Kimura模型[17]估算系统进化矩阵,用MEGA5.0软件采用邻接法[18]构建系统进化树.重复取样1000次进行自展值分析来评估系统进化树拓扑结构的稳定性.并定义16S rRNA基因序列相似性低于98%划分为不同的分类单元(OTUs)[14].克隆文库覆盖率(C)计算公式:C=[1-(n/N)]× 100%,n代表在克隆文库中OTUs的数量,N代表克隆文库的总数[19].本研究中得到的序列已提交GenBank,登录号为KM031411-KM031479.
1.2.5 数据处理 采用PAST软件对盐碱土中放线菌多样性指数:香农指数(Shannon)、辛普森指数(Simpson)、物种丰富度指数(Margalef)、均匀度指数(Evenness)和Chao-1指数进行计算分析.稀释性曲线(rarefaction curve)采用Analytic Rarefataction version 1.3软件进行分析.采用Office Excel 2003进行数据基本处理,用SPSS17.0统计软件对放线菌OTUs数和土壤理化因子间进行简单相关分析.
2 结果与分析
2.1 16S rRNA基因文库构建及多样性分析
经ARDRA酶切分析,将具有不同谱型的克隆进行测序,所测序列用Mothur软件进行嵌合体检验并去除嵌合体序列,从原生盐碱土(S1)、次生盐碱土(S2)和农田土(S3)样品基因文库中分别获得了22、36和11条有效序列,并将16S rRNA基因序列相似性低于98%作为划分不同OTUs的标准,S1、S2和S3 3个基因文库中分别含有20、32 和10个OTUs.多样性指数分析结果见表2.从稀释性曲线(图1)可以看出,S1、S2克隆文库的曲线均未呈现出趋于平缓的趋势.Kemp等[20]的研究结果表明,随着库容(Library size)的增大,OTUs的数目在增加,未有文库能够穷尽样品中微生物的多样性,这也说明S1、S2样品中放线菌多样性比较丰富,随着克隆数目增加,还会有新基因型出现.而S3样品克隆文库曲线已趋于平缓,库容已足够,基本上涵盖了该样品中放线菌的绝大多数物种.
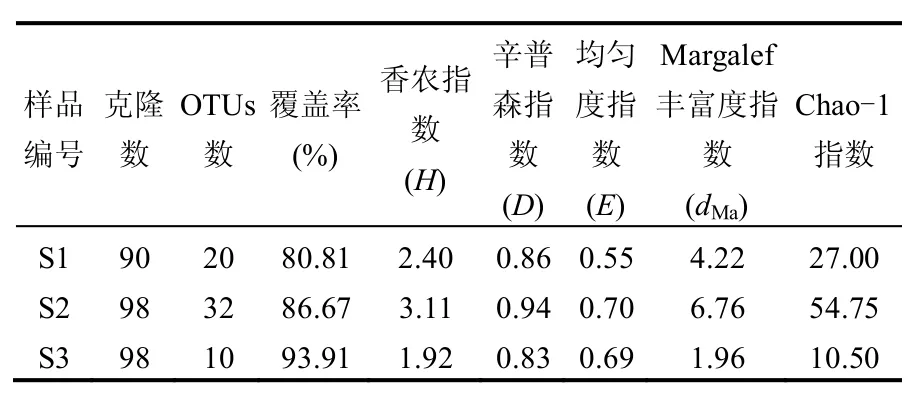
表2 河西走廊石羊河下游地区盐碱土中未培养放线菌多样性指数Table 2 Diversity index of uncultured actinomycetes at the saline-alkali soil in downstream area of Shiyang River of Hexi Corridor

图1 河西走廊石羊河下游地区盐碱土中未培养放线菌稀释性曲线Fig.1 Rarefaction curve of unculturable actinomycetes at saline-alkali soil in downstream area of Shiyang River of Hexi Corridor
2.2 系统发育分析
通过对河西走廊石羊河下游地区3种不同类型土壤中放线菌16S rRNA基因序列系统发育分析显示,原生盐碱土(S1)16S rRNA基因文库中的20个OTUs分属于微球菌亚目(Micrococcineae)的微球菌科(Micrococcaceae)、弗兰克氏菌亚目(Frankineae)的中村氏菌科(Nakamurellaceae)、丙酸杆菌亚目(Propionibacterineae)的类诺卡氏菌科(Nocardioidaceae)、链霉菌亚目(Streptomycineae)的链霉菌科(Streptomycetaceae)、棒杆菌亚目(Corynebacterineae)的棒状杆菌科(Corynebacteriaceae)和诺卡氏菌科(Nocardiaceae).另外,还有43.34%的克隆属于放线菌未知类群,它们可能代表了放线菌纲(Actinobacteria)和酸微菌纲(Acidimicrobineae)新科级别的未知类群.次生盐碱土(S2)16S rRNA基因文库中的32个OTUs分属于微球菌亚目(Micrococcineae)的纤维素单胞菌科(Cellulomonadaceae)和微球菌科(Micrococcaceae)、弗兰克氏菌亚目(Frankineae)的地嗜皮菌科(Geodermatophilaceae)、丙酸杆菌亚目(Propionibacterineae)的类诺卡氏菌科(Nocardioidaceae)、微单孢菌亚目(Micromonosporineae)的小单孢菌科(Micromonosporaceae)、假诺卡氏亚目(Pseudonocardineae)的伪诺卡氏菌科(Pseudonocardiaceae)、链霉菌亚目(Streptomycineae)的链霉菌科(Streptomycetaceae)、链孢囊菌亚目(Streptosporangineae)的链孢囊菌科(Streptosporangiaceae)和高温单孢菌科(Thermomonosporaceae)、动孢菌亚目(Kineosporiineae)的动孢囊菌科(Kineosporiaceae)、糖霉菌亚目(Glycomycineae)的糖霉菌科(Glycomycetaceae).并且,占19.39%的克隆属于放线菌的未知类群.在农田土(S3)16S rRNA基因文库中的10个OTUs分属于微球菌亚目(Micrococcineae)的微球菌科(Micrococcaceae)和博戈里亚湖菌科(Bogoriellaceae)、弗兰克氏菌亚目(Frankineae)的地嗜皮菌科(Geodermatophilaceae)和中村氏菌科(Nakamurellaceae)、丙酸杆菌亚目(Propionibacterineae)的类诺卡氏菌科(Nocardioidaceae).其中,仅有3.06%的克隆属于放线菌的未知类群.在3个不同土壤类型的基因文库中,微球菌亚目(Micrococcineae)所占比例最大,为3个基因文库的最优势类群.
2.3 土壤理化因子与放线菌数的简单相关分析
已有研究表明,土壤中放线菌数量与pH值有较为密切的关系[9],但该地区各类盐碱土壤的pH值非常接近,因此对放线菌数量影响不大.通过对放线菌OTUs数量与土壤理化因子间的相关分析(表3)可以看出,土壤有机质与土壤放线菌OTUs数呈显著负相关,相关系数为-0.697;土壤有效磷与土壤放线菌数呈现出极显著相关,该结果与本课题组之前在对河西走廊盐碱土壤中微生物数量与土壤理化因子关系的研究结果相一致,在理化因子中对放线菌影响最大的是速效磷,纤维素酶活性在一定程度上也会影响放线菌的分布[9].
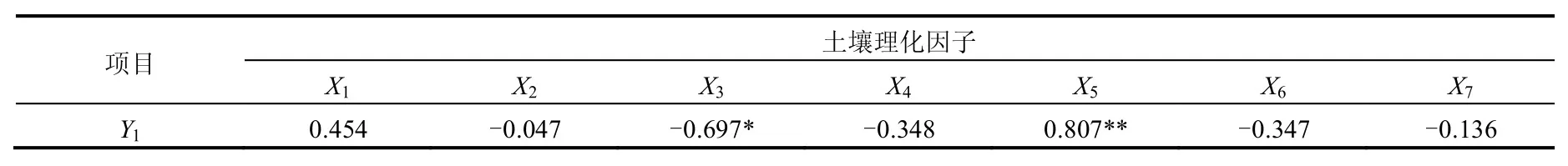
表3 土壤理化因子与放线菌数的简单相关分析Table 2 Simple correlation between actinomycetes quanity and soil physical and chemical factors
3 讨论
通过纯培养分离方法一直是人们开发利用微生物资源的直接手段,但是人工培养出来的微生物只占自然界的1%左右,而很多的微生物资源却因无法培养而不能被人类开发利用[21-22],同时也难以准确反映出土壤中微生物的多样性信息.采用基于16S rRNA基因的未培养技术以发现更多新的未培养微生物类群,使其能够更加真实的反映出土壤中微生物的生态分布情况[23-24].目前对于极端环境条件下的放线菌多样性研究较多.夏占峰等[25]对艾丁湖沉积物放线菌多样性的研究认为,该环境中优势放线菌为微球菌科的罗斯氏菌属(Rothia);姜怡等[26]采用DGGE、纯培养法,重点研究了新疆、青海及埃及的重盐碱环境的放线菌分布情况,种类组成,生物学特性.发现了1个新科,8个新属及30多个新种;关统伟等[27]对中国新疆硝尔库勒盐湖沉积物中放线菌的多样性进行了研究,所获得的51个克隆序列属中52.9%的克隆序列分布于放线菌门(phylum Actinobacteria)放线菌亚纲(Actinobacteridae)的链孢囊菌亚目(Streptosporangineae)、假诺卡氏亚目(Pseudonocardineae)、弗兰克氏菌亚目(Frankineae)、链霉菌亚目(Streptomycinea)和微球菌亚目(Micrococcineae)5个亚目和酸微菌亚纲(Acidimicrobidae)中;贾晓宇等[28]对新疆红井子盐碱土壤中的放线菌物种多样性研究认为,61个克隆序列分布于放线菌纲(Actinobacteria)的放线菌亚纲(Actinobacteridae)、酸微菌亚纲(Acidimicrobidae)和红色杆菌亚纲(Rubrobacteridae).本研究从河西走廊石羊河下游地区的原生盐碱土中获取得到放线菌目的微球菌亚目(Micrococcineae)、弗兰克氏菌亚目(Frankineae)、丙酸杆菌亚目(Propionibacterineae)、链霉菌亚目(Streptomycineae)、棒杆菌亚目(Corynebacterineae)5个亚目和放线菌未知类群;从次生盐碱土中得到了微球菌亚目(Micrococcineae)、弗兰克氏菌亚目(Frankineae)、丙酸杆菌亚目(Propionibacterineae)、微单孢菌亚目(Micromonosporineae)、假诺卡氏亚目(Pseudonocardineae)、链霉菌亚目(Streptomycineae)、链孢囊菌亚目(Streptosporangineae)、动孢菌亚目(Kineosporiineae)、糖霉菌亚目(Glycomycineae)共9个亚目和放线菌的未知类群;在农田土中获得了微球菌亚目(Micrococcineae)、弗兰克氏菌亚目(Frankineae)、丙酸杆菌亚目(Propionibacterineae)3个亚目和放线菌的未知类群.通过比较相关研究,在不同的生态环境体系中放线菌种群在分类地位和分布特征上有着明显差异.不同环境中放线菌种群在分类地位虽然有相似性,但彼此之间又有差异,可能是因为不同的生态环境体系决定了环境中微生物种群结构组成的不尽相同,土壤微生物群落组成与多样性也体现出明显地区特异性[29].另外,通过纯培养方法与分子生态学方法相结合进行不同环境中放线菌种群结构及多样性的研究,可使研究者能够更加真实准确的揭示出该环境中放线菌的种群组成及多样性.
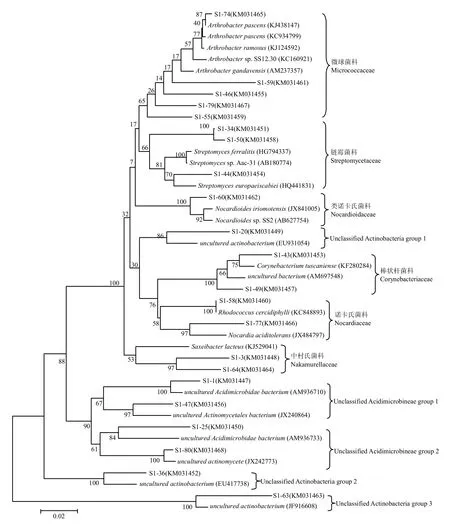
图2 基于原生盐碱土中放线菌16S rRNA 基因序列构建的系统进化树Fig.2 Phylogenetic tree of actinomycetes based on 16S rRNA gene sequences in primary saline-alkali soil
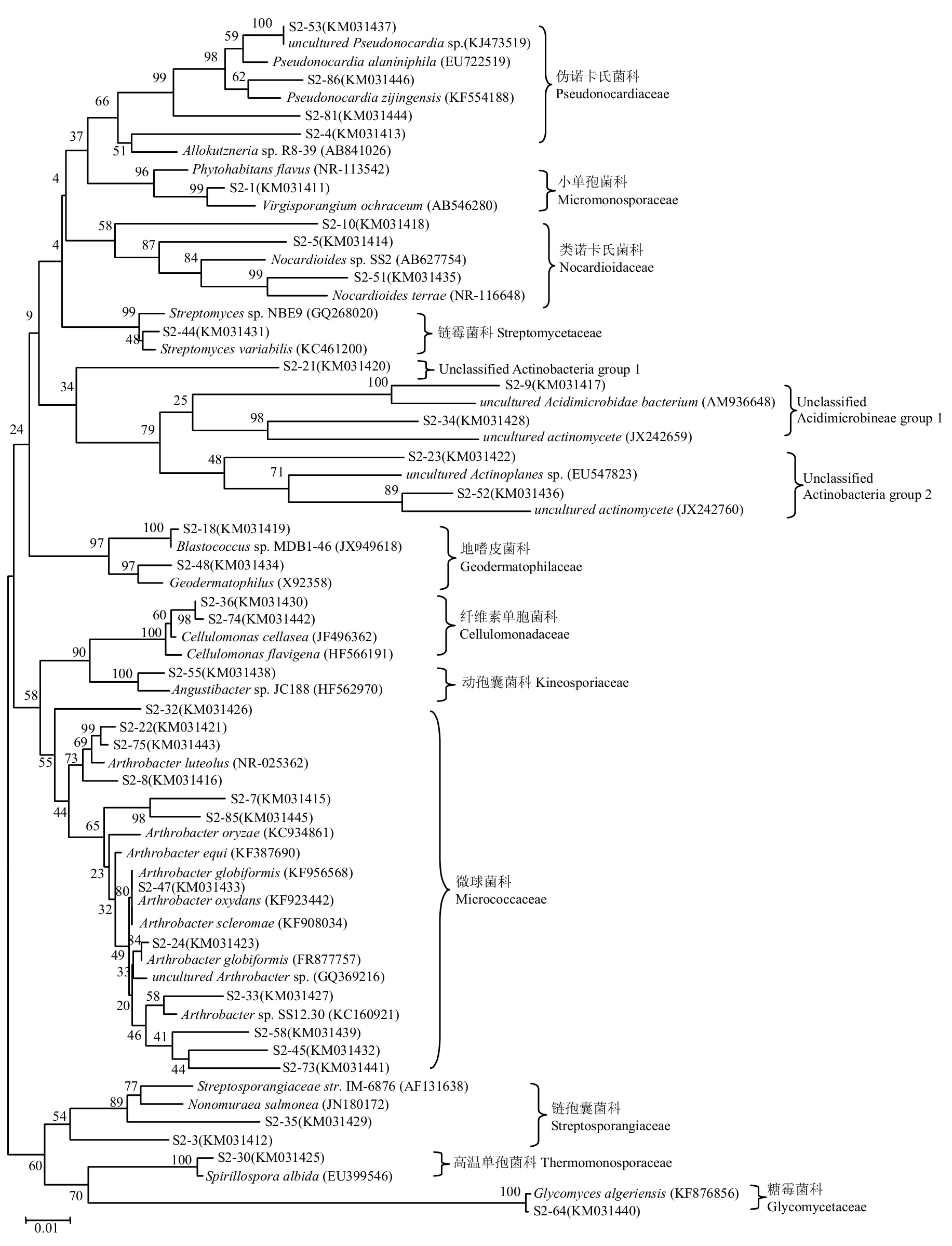
图3 基于次生盐碱土中放线菌16S rRNA 基因序列构建的系统进化树Fig.3 Phylogenetic tree of actinomycetes based on 16S rRNA gene sequences in secondary saline-alkali soil
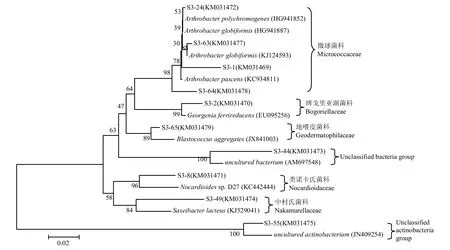
图4 基于农田土中放线菌16S rRNA 基因序列构建的系统进化树Fig.4 Phylogenetic tree of actinomycetes based on 16S rRNA gene sequences in farmland soil
4 结论
4.1 对河西走廊石羊河下游地区的3种不同类型土壤中放线菌多样性进行了初步研究,研究表明:原生盐碱土中放线菌分属于5个亚目6个科和放线菌未知类群;次生盐碱土放线菌分属于9个亚目11个科和放线菌的未知类群;农田土放线菌分属3个亚目5个科和未知类群.其中,微球菌亚目(Micrococcineae)是3种不同类型土壤中放线菌的优势种群.
4.2 多样性指数和稀释性曲线分析结果显示,3种不同类型土壤中放线菌多样性以次为次生盐碱土>原生盐碱土>农田土.
4.3 通过对放线菌OTUs数量与土壤理化因子间的相关分析可以看出,土壤有机质与土壤放线菌OTUs数呈显著负相关,土壤有效磷与土壤放线菌OTUs数呈现出极显著正相关.
[1]Bérdy J.Bioactive microbial metabolites[J].The Journal of Antibiotics,2005,58(1):1-26.
[2]Proksch P,Edrada R A,Ebel R.Drugs from the seas-current status and microbiological implications[J].Applied Microbiology and Biotechnology,2002,59(2/3):125-134.
[3]Wu S J,Fotso S,Li F,et al.Amorphane sesquiterpenes from a marine Streptomyces sp. [J].Journal of Natural Products,2007,70(2):304-306.
[4]刘全永,胡江春,薛德林,等.海洋微生物生物活性物质研究[J].应用生态学报,2002,13(7):90l-905.
[5]Hughes C C,Prieto-davo A,Jensen P R,et al.The marinopyrroles,antibiotics of an unprecedented structure class from a marine Streptomyces sp.[J].Organic Letters,2008,10(4):629-631.
[6]安登第,陈玉梅,李 进,等.银沙槐内生放线菌抗菌活性及其与内生细菌的拮抗关系[J].应用生态学报,2010,21(4):1021-1025.
[7]肖亚中.河西走廊盐碱土壤中的微生物[J].微生物学通报,2012,39(3):415.
[8]牛世全,杨婷婷,李君锋,等.盐碱土微生物功能群季节动态与土壤理化因子的关系[J].干旱区研究,2011,28(2):328-334.
[9]牛世全,杨建文,胡 磊,等.河西走廊春季不同盐碱土壤中微生物数量、酶活性与理化因子的关系[J].微生物学通报,2012,39(3):416-427.
[10]戴晟懋,邱国玉,赵 明.甘肃民勤绿洲荒漠化防治研究[J].干旱区研究,2008,25(3):319-324.
[11]岳东霞,杜 军,巩 杰,等.民勤绿洲农田生态系统服务价值变化及其影响因子的回归分析[J].生态学报,2011,31(9): 2567-2575.
[12]李小玉,肖笃宁,何兴元,等.内陆河流域中、下游绿洲耕地变化及其驱动因素—以石羊河流域中游凉州区和下游民勤绿洲为例[J].2006,26(3):671-680.
[13]Zhou J Z,Bruns M A,Tiedje J M.DNA recovery from soils of diverse composition[J].Applied and Environmental Microbiololgy,1996,62(2):316-322.
[14]Stach J E M,Maldonado L A,Ward A C,et al.New primers for the class Actinobacteria: application to marine and terrestrial environments[J].Environmental Microbiology,2003,5(10):828-841.
[15]Schloss P D,Westcott S L,Ryabin T,et al.Introducing mothur:open-source,platform-independent,community-supported software for describing and comparing microbial communities[J].Applied and Environmental Microbiology,2009,75(23):7537-7541.
[16]Thompson J D,Gibson T J,Plewniak F,et al.The CLUSYAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools[J].Nucleic Acids Research,1997,25(24):4876-4882.
[17]Kimura M.A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences[J].Journal of Molecular Evolution,1980,16(2):111-120.
[18]Saitou N,Nei M.The neighbor-joining method: a new method for reconstructing phylogenetic trees[J].Molecular Biology and Evolution,1987,4(4):406-425.
[19]Good I J.The population frequencies of species and the estimation of population parameters[J].Biometrika,1953,40(3/4):237-264.
[20]Kemp P F,Aller J Y.Bacterial diversity in aquaticand other environments: what 16S rRNA 1ibraries can tell us[J].FEMS Microbiology Ecology,2004,47(2):161-177.
[21]陈 灏,唐小树,林 洁,等.不经培养的农田土壤微生物种群构成及系统分类的初步研究[J].微生物学报,2002,42(4):478-483.
[22]Torsvik V,Ovreas L.Microbial diversity and function in soil:from genes to ecosystems[J].Current Opinion in Microbiology,2002,5(3):240-245.
[23]白 洁,李海艳,张 健,等.黄海西北部沉积物中细菌群落16S rDNA多样性解析[J].中国环境科学,2009,29(12):1277-1284.
[24]Kirk J L,Beaudette L A,Hart M,et al.Methods of studying soil microbial diversity[J].Journal of Microbiological Methods,2004,58(2):169-188.
[25]夏占峰,关统伟,阮继生,等.艾丁湖沉积物放线菌多样性[J].微生物学报,2011,51(8):1023-1031.
[26]姜 怡,李文均,徐 平,等.盐碱环境放线菌多样性研究[J].微生物学报,2006,46(2):191-195.
[27]关统伟,吴晋元,吴晓阳,等.硝尔库勒湖沉积物中非培养放线菌多样性[J].微生物学报,2008,48(7):851-856.
[28]贾晓宇,贺江舟,关统伟,等.新疆红井子盐碱土壤非培养放线菌多样性[J].微生物学通报,2012,39(5):606-613.
[29]Girvan M S,Bullimore J,Pretty J N,et al.Soil type is the primary determinant of the composition of the total and active bacterial communities in arable soils[J].Applied and Environmental Microbiology,2003,69(3):1800-1809.
Diversity of actinomycetes at the saline-alkali soils in downstream area of Shiyang River of Hexi Corridor——Illustrated by the case of Minqin County.
LI Hai-yun,NIU Shi-quan*,KONG Wei-bao,ZHU Xue-tai,ZHANG Ai-mei,DA Wen-yan,HAN Cai-hong,YAN Wei-ru,GENG Hui(College of Life Science,Northwest Normal University,Lanzhou 730070,China).China Environmental Science,2015,35(6):1805~1813
In order to understand the population structure and diversity of actinomycetes at saline-alkali soils in Shiyang River area of Hexi Corridor,the total DNA was extracted from three types soils(primary saline-alkali soil,secondary saline-alkali soil and farmland soil).Meanwhile,actinomycetes 16S rRNA gene clone libraries were constructed by amplified total DNA products with actinomycetes specific primers,then the positive clone products were digested by Hae Ⅲ and HhaⅠ,and the clones with different type of electrophoretic bands(Amplifed Ribosomal DNA Restriction Analysis,ARDRA)were sequenced,furthermore,the phylogenetic trees were built and diversity index was analyzed.The results demonstrated that the 90positive clones from the primary saline-alkali soil were attributed to 20 OTUs,belonged to Micrococcineae,Propionibacterineae,Corynebacterineae,Frankineae,Pseudonocardineae,Actinomycetales and unknown groups.The 98 positive clones from the secondary saline-alkali soil were attributed to 32 OTUs,belonged to Micrococcineae,Propionibacterineae,Corynebacterineae,Frankineae,Pseudonocardineae,Actinomycetales and unknown groups.The 98 positive clones from farmland soil were attributed to 10 OTUs,belonged to Micrococcineae,Propionibacterineae,Corynebacterineae,Frankineae,Pseudonocardineae,Actinomycetales and unknown groups.Micrococcineae was the dominant population in all type soils.The diversity index and Rarefaction curves analysis showed that actinomycetes diversity in the three different types soils was in the order secondary saline-alkali soil >primary saline-alkali soil >farmland soil.
Hexi Corridor;saline-alkali soil;clone library;actinomycetes diversity
X171.2
A
1000-6923(2015)06-1805-09
李海云(1989-),男,甘肃永靖人,西北师范大学硕士研究生,主要从事微生物多样性研究.发表论文5篇.
2014-10-31
国家自然科学基金资助项目(31260134,30960078)
* 责任作者,教授,sqniu@nwnu.edu.cn
