高浓度酶快速胶内酶解技术用于磷酸化蛋白质组学分析
2015-11-03刘芳等
刘芳等

摘 要 聚丙烯酰胺凝胶电泳是一项高效的蛋白质分离技术,通过与质谱技术联用,可以鉴定成千上万的蛋白质点。但是,复杂繁琐的操作流程限制了它在蛋白质组学研究方面的广泛应用。本研究表明,高浓度的胰蛋白酶并不会影响胶内酶解后磷酸化肽段的富集,反而可以实现快速高效的蛋白酶解,据此建立了一种快捷、高效的蛋白质酶解方法。首先, 通过聚丙烯酰胺凝胶电泳,对50 μg复杂蛋白样品进行分离,分成5个组分; 然后, 选择高浓度胰蛋白酶酶解30 min; 最后,运用固定钛离子亲和色谱小柱离心法富集磷酸化肽段。经液相色谱-串联质谱分析,成功鉴定到约2000个磷酸化位点,而传统方法则只鉴定到不足1500个位点。实验表明,在高浓度胰蛋白酶的促进作用下,胶内酶解过程在0.5 h内即可以完成,并且得到比对照组更高的磷酸化位点的鉴定量和更低的酶解漏切率,而传统方法需要16 h。这也证实了本方法不仅可以加速酶解反应,同时还能提高蛋白酶解效率。
关键词 高浓度酶; 胶内酶解; 磷酸化蛋白质组学
1 引 言
聚丙烯酰胺凝胶电泳是一项高效的蛋白质分离技术,它可以实现包含有成千上万个蛋白质的复杂蛋白样品的深度分离[1]。通过与质谱技术的联用,可以实现这些蛋白质点的规模化鉴定,已经成为Bottom-up蛋白质组学研究方法(即“鸟枪法”)的重要组成部分。与多维液相色谱-质谱法相比,聚丙烯酰胺凝胶电泳-质谱法具有独特的优点:首先,通过聚丙烯酰胺凝胶电泳实现了蛋白混合物按照分子量大小的预分离,大大降低了样品的复杂度,从而可以获得高动态范围的蛋白混合物的鉴定分析; 其次,它对于盐离子、缓冲溶液以及表面活性剂的耐受程度较高,而这些物质都不利于后续质谱检测[2,3]。但是,它复杂、繁琐的操作流程和较低的酶解效率制约了其在蛋白质组学研究方面的进一步应用[4]。这主要是因为经电泳预分离后的蛋白,在后期的胶内酶解过程中,要经过多步的洗涤、缩干以及过夜酶解等操作,直接导致了该方法的通量低; 同时, 酶扩散效率低也是导致胶内酶解效率不高的一个主要原因。因此,很多研究者致力于研究快速高效的胶内酶解方法,以满足蛋白质组学研究对样品预处理阶段日益增强的要求。
为了提高胶内酶解效率,Havlis等[5]研究了胶内酶解反应的动力学过程,考察了反应温度、酶浓度、酶解时间以及蛋白条带的预酶解对于整个酶解过程的影响。他们通过动力学数据优化了酶解反应条件,发现选择较高的酶解温度、较高的酶浓度,在不影响酶解效率的前提下,可在30 min内实现快速酶解,这主要得益于他们使用了甲基化处理后的胰蛋白酶,此种酶的自酶解概率低,并且可以耐受比较高的酶解温度,却不影响最终的酶解特异性。但是,该方法并没有得到广泛应用,可能是因为这种酶价格昂贵,难以推广使用。在此基础之上,Dycka等[6]研究了超声处理和红外辐射对于胶内酶解的促进作用,证实了这两种方法都是潜在的可以用来提高胶内酶解效率的策略,可以使原本耗费过夜的酶解过程在几分钟内轻松实现。除此之外,微波辅助法也已经是一种被广泛用于加速胶内酶解的方法,但是将它用于复杂蛋白的快速胶内酶解还存在一定的难度[7~9]。Saveliev等[3]则利用质谱兼容的表面活性剂提高胶内酶解效率,蛋白酶解加上肽段提取的总时间不超过1 h,并且,肽段的回收率也高于传统方法,显示出了这种表面活性剂的独特应用,只是这种表面活性剂在酶解过程中会发生降解,其降解产物对于质谱鉴定的影响仍亟需进一步的考察。
众所周知,酶解过程中,使用高浓度的胰蛋白酶,蛋白的酶解速率也就随之增高[10]。这是一种很直接的用于加速蛋白酶解的方法,但是却存在严重的胰蛋白酶自酶解现象。高强度的胰蛋白酶自酶解峰,会在质谱分析时抑制正常肽段出峰,这也正是蛋白酶解时不宜选用过高酶浓度的重要原因。本研究发现,高浓度的酶对后续磷酸化的富集分析几乎没有影响。这是因为只有磷酸肽才能被富集出来进行后续质谱分析,从而避免了胰蛋白酶自酶解现象对于后续质谱检测的影响。本研究将该方法应用于磷酸化蛋白质组学的研究。在高浓度胰蛋白酶的促进作用下,胶内酶解反应可在0.5 h内基本完成,而在传统方法中,这一过程常需要16 h左右。继而运用填充了固定钛离子亲和色谱(Ti-IMAC)材料的小柱(Tip)进行磷酸肽富集,于50 μg子宫颈癌细胞(HeLa)蛋白样品中,成功鉴定到了约2000多个磷酸化位点,比传统方法多鉴定到了约40%的磷酸化位点; 酶解漏切率却远低于传统方法。这说明此方法不仅可以加速酶解反应,同时还能提高胶内酶解效率。不仅如此,本方法在高分子量蛋白和低丰度蛋白的鉴定方面,也都明显优于传统方法,进一步显示出了本方法相较于传统方法良好的互补性。
2 实验部分
2.1 仪器与试剂
LTQ Orbitrap Velos质谱仪(美国Thermo公司),配有纳升级电喷雾离子源和六通阀; Accela 600 HPLC液相色谱系统(美国Thermo公司),配有脱气机和四元液相泵。
子宫颈癌细胞(HeLa)来源于中国医学科学院血液病研究所; RPMI-1640细胞培养基(美国Gibco Invitrogen公司); 甲酸(FA)、三氟乙酸(TFA)、乙二胺四乙酸二钠(EDTA)、乙二醇双(2-氨基乙基醚)四乙酸(EGTA)、蛋白酶抑制剂(Cocktail)、NaF、正钒酸钠(Na3VO4)、四甲基乙二胺(TEMED)、过硫酸铵(AP)、二硫苏糖醇(DTT)、碘代乙酰胺(IAA)和TPCK 处理的胰蛋白酶(美国Sigma 公司); 乙腈(色谱纯)、25%氨水(德国Merck公司); 40%丙烯酰胺-甲叉双丙烯酰胺(29∶1)、考马斯亮蓝R250(美国Bio-Rad公司); 预染蛋白分子量标准、蛋白上样缓冲液(美国Thermo公司); GE Loader tips (20 μL, 德国Eppendorf公司); 10% SDS(上海碧云天生物技术研究所); 二次去离子水经过Mill-Q水纯化系统处理; 其余试剂均为分析纯。
2.2 实验方法
HeLa细胞在RPMI 1640培养基内培养, 加入了10%新生小牛血清和100 U/mL双抗,温度设定为37℃,CO2浓度为5%。当细胞长至对数生长期后,收集细胞并用冷的PBS缓冲溶液洗涤两次。采用文献\[11\]的方法进行蛋白提取。
将已知浓度的蛋白溶液和电泳上样缓冲液按照一定比例混合之后,沸水浴5~10 min。将适量蛋白样品上样到12%聚丙烯酰胺凝胶中,在80 V电压下,浓缩20 min,继而调至120 V,继续电泳至蛋白充分分离。将所得胶条用考马斯亮蓝溶液染色,脱色至看到清晰的蛋白条带。扫描完蛋白条带之后,将蛋白条带分成多个组分,手动切割成1 mm× 1 mm的胶条,并转移至离心管内,加入脱色液(100 mmol/L NH4HCO3-乙腈(1∶1, V/V), 在4℃条件下,振荡脱色,30 min换一次脱色液,至充分脱去考马斯亮蓝的颜色。加入纯乙腈使胶条充分缩干,继而加入20 mmol/L DTT溶液,37℃振荡反应2 h; 洗去多余DTT溶液后,加入55 mmol/L IAA溶液,室温黑暗中振荡反应1 h。充分洗涤剩余的IAA溶液,用乙腈缩干胶条,实验组加入300 ng/ 的胰蛋白酶溶液,而对照组则是加入15 ng/ 的胰蛋白酶溶液,放置于4℃,使胶条充分吸收酶溶液至饱和状态,再稍微补加20~30 μL 100 mmol/L NH4HCO3溶液至液面浸过胶条以上。实验组在37℃酶解反应0.5 h,而对照组在37℃酶解反应16 h。将所得肽段萃取溶液,在室温真空干燥,用80%乙腈-6%三氟乙酸溶液复溶,继而用填有Ti-IMC材料的Tip进行磷酸化肽段的富集。所得磷酸化肽段,室温真空干燥,用0.1%甲酸溶液复溶,用于质谱上样。
具体富集过程同文献[12,13]一致。将GE Loader tips(20 μL)的尖端塞入少许脱脂棉,充当塞子。取适量Ti-IMAC材料(材料与蛋白的质量比为15∶1),用上样缓冲液(80% ACN, 6% TFA)重悬,加入枪头内,离心去掉溶液,待用。蛋白酶解液用上样缓冲液复溶后,加入枪头内,以1000 g的转速离心富集。依次用洗涤缓冲液1(50% ACN, 6% TFA, 200 mmol/L NaCl)和2(30% ACN, 0.1% TFA)分别洗涤一次,保持转速为1500 g。最后,用100 μL 10%氨水洗脱磷酸肽两次,合并洗脱液,室温冻干, -30℃保存待用。
2.3 质谱方法
复溶的样品采用自动进样系统上样至质谱仪,自动上样系统包括一段4 cm的毛细管富集柱(200 μm i.d.)和一段15 cm的毛细管分析柱(75 μm i.d.),将毛细管分析柱在高温下拉成内径约为3 μm的尖头,用气压法将两根柱子都填充C18 AQ填料。液相色谱-质谱系统的流速约为200 nL/min。流动相A为0.1% 甲酸-水溶液; 流动相B 为0.1% 甲酸-乙腈溶液。洗脱梯度设置如下: 0~2 min,0%~2% B; 2~92 min,2%~25% B; 92~97 min,25%~35% B; 97~100 min,35%~80% B; 100~110 min,80% B; 110~120 min,100% A。
LTQ Velos质谱仪在正离子模式下进行肽段检测,离子传输毛细管的温度为250℃,喷雾电压为2.2 kV,归一化碰撞能量为35.0%。采用数据依赖模式(DDA)进行采集,包含一次MS全扫描(m/z 400~2000),分辨率为60000,然后对其中强度最高的20个峰进行二级MS/MS扫描分析,系统的操作和数据的采集都使用Xcalibur软件(v2.1,Thermo公司)完成。
2.4 数据处理
所收集的*.RAW文件用Thermo Proteome Discoverer Daemon (v1.4)转换成*.MGF格式,通过Mascot Daemon(Version 2.3)在人源蛋白质数据库(Uniprot human,包含88473个条目)进行检索。搜库参数设置如下:胰蛋白酶酶切,允许最多两个漏切位点,固定修饰为半胱氨酸碘代酰胺烷基化(+57.0215 Da),可变修饰为甲硫氨酸的氧化(+15.9949 Da),丝氨酸、苏氨酸、酪氨酸的磷酸化(+79.96633 Da)。母离子的质量容忍偏差为10 ppm,碎片离子为0.8 Da。导出肽段时设置Score>25、p<0.01,同时控制假阳性率(FDR)<1%。
3 结果与讨论
3.1 实验条件的选择
动态范围宽和化学计量数低一直是磷酸化蛋白质组学深入研究的制约因素,这对样品的预处理过程提出了一定的要求,尤其是微量样品的磷酸化分析。在之前的工作中,本研究组发展了一种便捷高效的样品预处理方法用于磷酸化蛋白质组学分析[14]。该方法利用高浓度胰蛋白酶可以促进蛋白酶解这一原理,实现了短时间内的样品处理; 同时利用Ti-IMAC Tip离心法富集磷酸化肽,降低样品的损失,最终实现微量样品的快速磷酸化分析。Ti-IMAC Tip离心法就是将适量Ti-IMAC材料填充于特制的枪头内,运用离心力进行磷酸化肽段的富集。该方法简单高效,同时可以减少样品的损失,适用于微量样品的组学分析。本研究将此方法与高分辨的SDS-PAGE技术联用,旨在建立高效快速的胶内酶解方法,并用于磷酸化蛋白质组的分析。
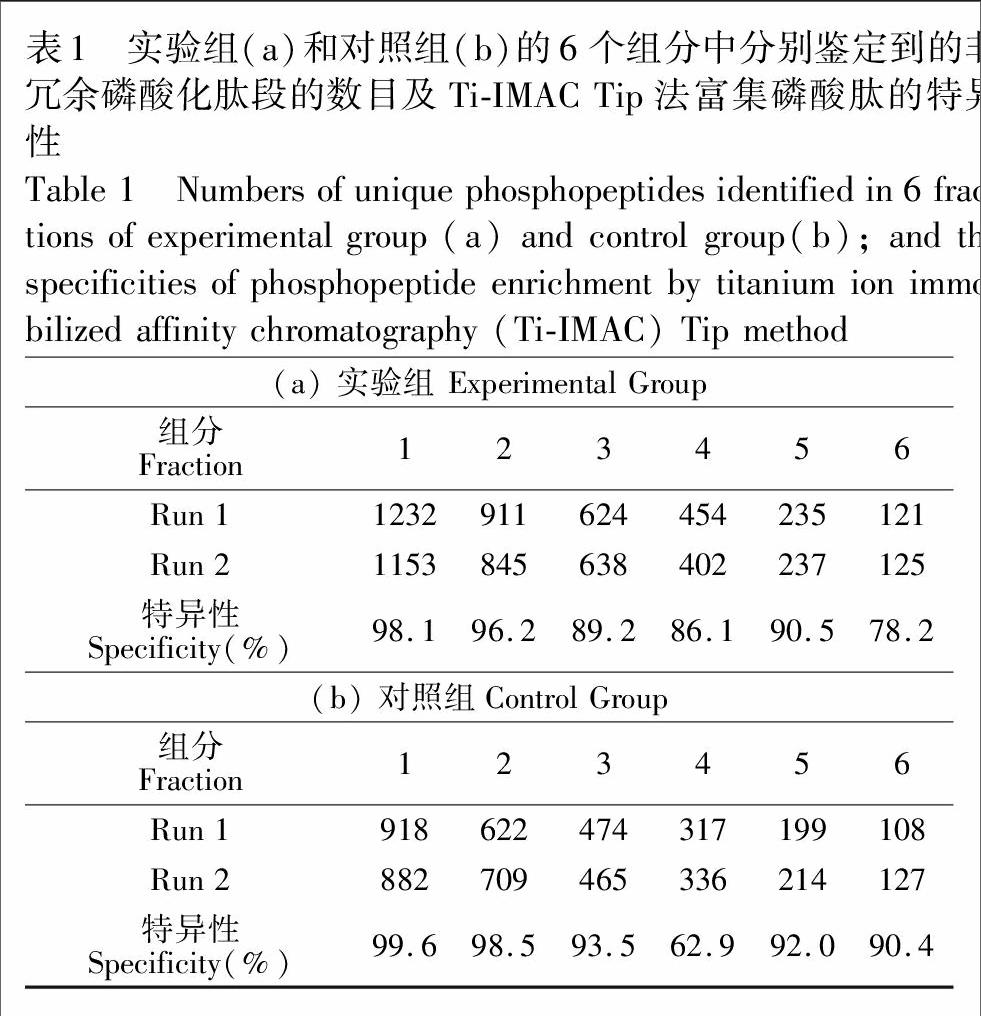
研究表明,选用高浓度胰蛋白酶酶解对于后续磷酸化肽段的富集几乎没有影响,甚至当酶量增大至蛋白量的10倍时,依然可以鉴定到相同数量的磷酸化位点[14]。因此,可以采用高浓度胰蛋白酶加速胶内酶解反应,并将其应用于磷酸化蛋白质组学研究。首先,基于此前研究的酶解反应条件,验证此方法的可行性。如图1A所示,将单一泳道上的蛋白条带(含120 μg HeLa 蛋白)依据主蛋白条带(即颜色最深的条带)分成6个组分。取相邻两个泳道同时进行实验组和对照组的实验。实验组采用文献[14]优化得到的酶解反应条件,即酶浓度为300 ng/μL,酶解时间30 min; 而对照组则采用传统的酶解方法,所用的酶浓度为15 ng/μL,酶解时间为16 h。待酶解完成后,用Ti-IMAC Tip法富集酶解液中的磷酸化肽段,而后进行LC-MS/MS分析,重复一次。结果如表1所示,在每一个组分里,实验组都获得了比对照组更高的磷酸肽的鉴定量,并且所有组分的磷酸肽的富集特异性基本稳定在80%以上,证明了Ti-IMAC Tip法的高效性和稳定性,也同时验证了这种快速高效的酶解方法的可行性。
3.2 微量样品的磷酸化分析
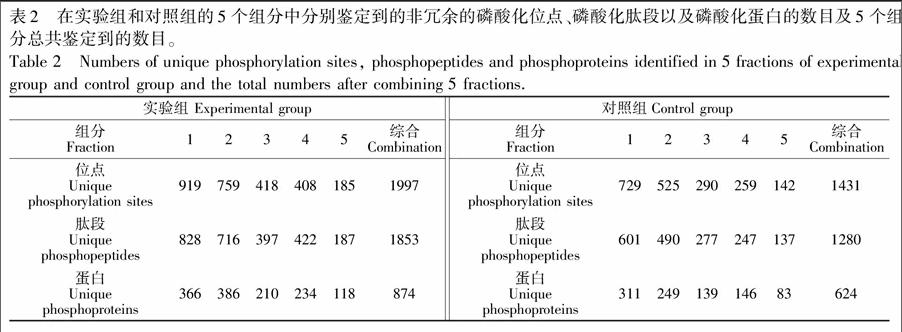
将此方法应用于微量样品的磷酸化分析。如图1B,将含有50 μg HeLa蛋白的胶条分成5个组分,酶解富集后,进行LC-MS 分析。质谱结果如表2所示,相较于对照组,实验组的每一个组分在短短30 min内都鉴定到了更多非冗余的磷酸化位点、磷酸化肽段以及磷酸化蛋白。将5个组分的鉴定结果综合在一起,对照组最终鉴定到1431个磷酸化位点,而实验组则鉴定到多达1997个磷酸化位点,比对照组多鉴定到约40%的磷酸化位点。同样,在磷酸化肽段鉴定方面,实验组比对照组多鉴定到约45%的磷酸化肽段; 在磷酸化蛋白鉴定方面,实验组则比对照组多鉴定到40%的磷酸化蛋白。3个方面的数据都说明,本方法获得了更好的质谱鉴定结果,并且将原本复杂繁琐的胶内酶解过程缩短至仅需30 min。
3.3 磷酸化肽段漏切率的比较
酶解漏切率的高低是检验蛋白酶解是否充分高效的重要指标之一,而且高漏切率会增加蛋白搜库匹配的错误率,从而影响蛋白的可靠鉴定。本研究统计了实验组和对照组中酶解反应的漏切率(漏切率=含有漏切位点的磷酸肽数目/所有的磷酸肽数目)。结果如图2所示,在5个组分中,对照组的酶解漏切率都明显高于实验组。已知在磷酸化蛋白质组学分析中,影响蛋白酶解漏切的主要因素是磷酸化修饰残基和其它一些酸性氨基酸(天冬氨酸、谷氨酸)离酶切位点(精氨酸、赖氨酸)太近,以至阻碍了胰蛋白酶识别位点的效率[15]。因此,有文献指出,磷酸化肽段的漏切率高达普通肽段的4倍,而本实验鉴定到的漏切率与文献[6,16]报道基本一致。进一步分析可以得知,实验组的漏切率明显低于对照组,极有可能是因为高浓度胰蛋白酶的存在减弱了磷酸化修饰残基和其它酸性氨基酸对蛋白酶切的影响。这也证明了高浓度的胰蛋白酶在加速酶解反应的同时,也可以提高蛋白酶解的效率。
3.4 相邻组分间磷酸化蛋白鉴定的覆盖度
统计了相邻组分间磷酸化蛋白鉴定的覆盖率。如图3所示,无论是实验组还是对照组,两个相邻组分间所鉴定到的磷酸化蛋白的覆盖率均约为20%,这充分证明了聚丙烯酰胺凝胶电泳法优异的分离蛋白的能力,也是聚丙烯酰胺凝胶电泳法不可被取代的重要原因之一。在实验组和对照组中,
分析了不同组分所鉴定到的所有蛋白的平均分子量的分布情况。结果如图4所示,蛋白平均分子量依次递减的趋势是与SDS-PAGE分级的情况完全一致的,这也再次从侧面证明了相邻组分间的覆盖率是较低的,几乎不会影响到相邻组分的蛋白鉴定。此外,本研究还发现,实验组鉴定到的蛋白的平均分子量比对照组略高,这表明了实验组在鉴定高分子量蛋白方面似乎更有优势。这可能是由于大蛋白体积较大、酶切位点较多,因而,与胰蛋白酶接触的几率也就较高,在较短的酶解时间内更容易被鉴定到,这也说明了本方法与传统方法具有一定的互补性。计算了本方法和传统方法之间的覆盖率,结果为42.7%。该数值与文献\[17\]所报道的不同方法间的覆盖率值是相接近的, 说明本方法是可靠的,可以对传统方法进行一定程度的补充。
3.5 磷酸化蛋白丰度的差异
对实验组和对照组中所鉴定到的蛋白丰度的分布情况进行了统计和比较。所有文献已报道的蛋白丰度值[18]都可以在线查询获得(http://pax-db.org/), 将得到的蛋白丰度值取对数,得丰度分布图。如图5所示,在低丰度蛋白的鉴定方面,实验组表现出更高的鉴定效率; 而在高丰度蛋白的鉴定方面,对照组则表现出较高的鉴定效率。正如文献\[19\]讨论的,在整个酶解过程中,最先发生酶解的是那些与酶结合力强,也就是较易断裂的位点,这是通过胰蛋白酶识别位点附近的序列实现的。当选择较短的酶解时间时,无论是高丰度的蛋白还是低丰度的蛋白,都是序列中最先被切割而被释放出来的肽段,与此同时,因为采用了高浓度的胰蛋白酶,使得低丰度的蛋白较快酶解完全,这样,在短时间内,低丰度蛋白产生的肽段就不易被高丰度蛋白产生的肽段所抑制; 而在低浓度的胰蛋白酶条件下,随着酶解时间的逐渐增长,所有蛋白的所有肽段都得到了释放,这样高丰度蛋白就会大大抑制低丰度蛋白,从而使低丰度蛋白的鉴定变得比较困难。众所周知,低丰度蛋白的鉴定一直是蛋白质组学研究的难点,而高浓度胰蛋白酶辅助酶解法在这方面体现出了一定的优势,因此,基于高浓度胰蛋白酶的快速酶解法具有更广阔的应用前景。
4 结 论
本研究建立了一种快速高效的胶内酶解方法,并将其应用于微量样品的磷酸化分析。通过采用高浓度的胰蛋白酶进行胶内酶解,在30 min内完成酶解反应,将其用于50 μg HeLa 蛋白的磷酸化分析,获得了远优于对照组的鉴定结果,并且酶解漏切率却低于传统方法。这证明了本方法的高效性,不仅可以促进胶内酶解反应,同时可以提高胶内酶解的效率。除此之外,本方法在鉴定高分子量、低丰度蛋白方面,都显示出了优于传统方法的性能,本方法可以作为传统方法的补充,会获得更广阔的应用空间。
References
1 Grg A, Weiss W, Dunn M J. Proteomics, 2004, 4(12): 3665-3685
2 Shevchenko A, Tomas H, Havlis J, Olsen J V, Mann M. Nat. Protoc., 2007, 1(6): 2856-2860
3 Saveliev S V, Woodroofe C C, Sabat G, Adams C M, Klaubert D, Wood K, Urh M. Anal. Chem., 2012, 85(2): 907-914
4 Rabilloud T, Vaezzadeh A R, Potier N, Lelong C, Leize-Wagner E, Chevallet M. Mass Spectrom. Rev., 2009, 28(5): 816-843
5 Havli J, Thomas H, ebela M, Shevchenko A. Anal. Chem., 2003, 75(6): 1300-1306
6 Dycka F, Bobal P, Mazanec K, Bobalova J. Electrophoresis, 2012, 33(2): 288-295
7 Pramanik B N, Mirza U A, Ing Y H, Liu Y H, Bartner P L, Weber P C, Bose A K. Protein Sci., 2002, 11(11): 2676-2687
8 Juan H F, Chang S C, Huang H C, Chen S T. Proteomics, 2005, 5(4): 840-842
9 Sun W, Gao S, Wang L, Chen Y, Wu S, Wang X, Zheng D, Gao Y. Mol. Cell. Proteomics, 2006, 5(4): 769-776
10 Hustoft H K, Reubsaet L, Greibrokk T, Lundanes E, Malerod H. J. Pharm. Biomed. Anal., 2011, 56(5): 1069-1078
11 Bian Y, Ye M, Song C, Cheng K, Wang C, Wei X, Zhu J, Chen R, Wang F, Zou H. J. Proteome Res., 2012, 11(5): 2828-2837
12 Zhu J, Wang F, Chen R, Cheng K, Xu B, Guo Z, Liang X, Ye M, Zou H. Anal. Chem., 2012, 84(11): 5146-5153
13 Zhou H, Ye M, Dong J, Corradini E, Cristobal A, Heck A J R, Zou H, Mohammed S. Nat. Protoc., 2013, 8(3): 461-480
14 Liu F, Ye M, Pan Y, Zhang Y, Bian Y, Sun Z, Zhu J, Cheng K, Zou H. Anal. Chem., 2014, 86(14): 6786-6791
15 Molina H, Horn D M, Tang N, Mathivanan S, Pandey A. Proc. Natl. Acad. Sci. U. S. A., 2007, 104(7): 2199-2204
16 Lin Y, Li Y, Liu Y, Han W J, He Q Z, Li J L, Chen P, Wang X C, Liang S P. Electrophoresis, 2009, 30(20): 3626-3635
17 Bodenmiller B, Mueller L N, Mueller M, Domon B, Aebersold R. Nat. Methods, 2007, 4(3): 231-237
18 Wang M, Weiss M, Simonovic M, Haertinger G, Schrimpf S P, Hengartner M O, von Mering C. Mol. Cell. Proteomics, 2012, 11(8): 492-500
19 Ye M L, Pan Y B, Cheng K, Zou H F. Nat. Methods, 2014, 11(3): 220-222
