基于重组工程法高转化效率的大肠杆菌BL21DE3表达菌株构建
2015-10-21张飞飞石牡丹尚广东
张飞飞 石牡丹 尚广东


摘要 [目的]提高最常用的异源蛋白表达宿主菌E. coli BL21(DE3)的转化效率。[方法]利用基于质粒的重组工程系统和双链断裂修复功能。[结果]敲除了编码降解外源DNA的I型限制性内切酶的hsdR基因,获得了高转化效率以及其他功能与BL21(DE3)仍保持一致的LS1928菌株。[结论]获得的LS1928菌株可能会作为获得催化用酶的关键材料,进而在蛋白质工程等研究领域有着重要的作用。
关键词 重组工程;基因敲除;双链断裂修复;大肠杆菌BL21(DE3)
中图分类号 S188 文献标识码 A 文章编号 0517-6611(2015)20-029-03
Abstract [Objective] The aim was to improve the conversion efficiency of the most commonly used heterologous protein expression host strain. [Method] Type I restriction endonuclease capable of degrading exogenous DNA coding gene hsdR was knocked out via plasmidbased recombineering system as well as its inherent double strand break repair(DSBR) function. [Result]The obtained strain LS1928 was still consistent with E. coli BL21(DE3) in every aspect except higher conversion efficiency.[Conclusion] The newly developed strain has the potential to be used as key material to obtain catalytic enzyme and hence plays an important role in protein engineering.
Key words Recombineering; Gene knock out; DSBR; E. coli BL21(DE3)
酶是工业化生产的关键材料,也是现代绿色经济和工业生物技术的基础。野生型的酶常常难以具有人们所期望的最佳活性。高催化活性以及高催化专一性酶的获得常常依赖于基因突变,由于突变的随机性,目标突变只占最终突变的极小比例,一般在百万分之一的级别,因此高转化效率的菌株是获得高覆盖率蛋白突变体库的基础之一。突变体库也是研究基因或蛋白结构和功能的关键手段。
常规的得到蛋白突变体的试验步骤是将通过特定方法(如DNA shuffling[1]和易错PCR[2])所获得的突变体基因库以限制性内切酶进行酶切后所得的DNA片段和同样酶切的载体在T4连接酶的作用下相连,连接液转化至克隆宿主菌大肠杆菌(Escherichia coli)如DH10B或JM109,培养所得抗性菌株,从中提取质粒后,再转化至异源蛋白表达宿主菌。其中E.coli BL21(DE3)是最为常用的宿主菌,其他菌株一般为此菌株的衍生菌株。采用提取质粒再转化BL21(DE3)的两步法而非直接转化BL21(DE3)的原因在于后者的转化效率较低,一般低10倍以上。低效率使得获得目的突变的几率大大降低而不被采用。两步转化法不仅增加了试验步骤,且造成了一定量的(也可能是非常关键的)突变体库的损失。
重组工程的基本原理是利用重组酶催化同源片段之间发生重组,而进行DNA克隆和修饰的基因工程技术[3-4]。由于可以直接采用PCR扩增的中间含有抗性基因的线性同源片段(而无需将片段克隆至载体)进行转化,因此重组工程方法高效、简便和快捷。该研究所使用的重组酶基因是目前所广泛使用的,来源于λ噬菌体的Red重组系统,包括exo、bet和gam基因。Exo蛋白[5]是一种5′-3′DNA外切酶,结合在双链DNA的末端得到3′端突出的DNA分子;Bet蛋白[6]结合在突出的单链DNA分子上防止单链DNA被宿主单链核酸酶降解,同时还具有介导互补单链DNA退火;Gam蛋白[7]可与RecBCD结合抑制其降解DNA的活性。
BL21(DE3)基因组上hsdR基因编码I型限制性内切酶,在大肠杆菌细胞中起到一种“抗体”的作用,对外来的DNA有严格的限制。hsdR基因的变异或缺失会导致菌株细胞内的I型限制性内切酶活性缺失,这对外来基因的导入及质粒转化是有利的[8]。笔者通过利用重组工程的方法将E.coli BL21(DE3)菌株中的hsdR基因敲除,获得高转化效率的重组工程菌株。
1 材料与方法
1.1 材料
1.1.1 菌株與质粒。
E.coli DH10B、BL21(DE3)、MG1655、BW25141为江苏省麻醉学重点实验室保存的菌株。pET30a(+)和pBluescript II KS(-)为江苏省麻醉学重点实验室保存的质粒,pTKRed由美国Princeton大学Thomas Kuhlman教授惠赠。
1.1.2 试剂和仪器。
限制性内切酶、T4连接酶、IPTG、质粒提取试剂盒、胶回收试剂盒、PCR产物纯化试剂盒以及DNA分子量标准购自大连宝生物公司;PCR引物、蛋白质分子量标准、40%丙烯酰胺溶液、抗生素、L阿拉伯糖等购自上海生工公司;其余试剂均为国产分析纯。PCR仪为BioRad公司的S1000,电转化仪为BioRad公司的Gene PulserII。
1.1.3 DNA测序。
由南京思普金生物科技有限公司完成。
1.2 方法
1.2.1 分子生物学常规操作。
大肠杆菌的培养、PCR扩增、质粒提取和酶切鉴定以及十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDSPAGE)等试验按手册[9]进行。表达重组酶的电转化感受态的制备、DNA的电转化以及筛选等步骤均采用文献[10]的方法。 研究所使用的引物为:
1.2.2 质粒的构建。
1.2.2.1 卡那霉素两侧为ISceI酶切位点,R6K复制子质粒的构建。
以pKD4为模板,分别以R1115R1102和R1103R1116为引物,扩增得到0.3和0.6 kb片段,然后分别以ClaIPstI 和PstIXbaI酶切,通过三片段连接法克隆到pBluescript II KS(-)的ClaIXbaI位点上。经酶切和测序正确后,0.9 kb的ClaIXbaI酶切片段克隆到pKD4的相同位点得到pLS1662。pLS1662使用R6K复制子,以含有Pir基因的BW25141为宿主菌,这样以之为模板所扩增的片段转化到常规的大肠杆菌如MG1655就不会有质粒背景的干扰。
1.2.2.2 大肠杆菌醛酸酶yidS表达载体的构建。
以MG1655 基因组DNA为模板,AL1AL2扩增的0.9 kb醛缩酶yidS基因BamHIHindIII酶切后克隆至pET30a(+)/BamHIHindIII获得pLS182,酶切和测序正确。
1.2.3 基因敲除。
根据Kolisnychenko等的报道[10],综合运用重组工程和DSBR法进行基因敲除的基本步骤如下:首先将hsdR基因上下游各500 bp的同源片段以及两侧含有18 bp ISceI酶切位点的卡那霉素抗性基因通过重叠延伸PCR融合在一起,此线性融合片段还包含下游500 bp同源片段之后的50 bp的序列。将此线性融合片段电转化至表达重组酶的BL21(DE3),由重组酶催化同源片段之间的同源重组而将线性融合片段取代BL21(DE3)基因组上的hsdR基因部分,hsdR基因被敲除。随后经诱导表达的ISceI作用于所得菌株基因组中的ISceI酶切位点,引起基因组双链的断裂。此时菌株利用自身的同源重组系统如recA和recBCD催化线性整合片段和500 bp的同源片段下游的2个50 bp片段之间发生同源重组,即将2个片段之间的序列以及其中的1个片段去除,经过此步骤,修复了基因组双链的断裂,菌株恢复生长,最终得到hsdR基因敲除,且不含有任何的外源碱基序列的基因工程菌株,具体流程如图1所示。
2 结果与分析
2.1 BL21(DE3)菌株基因组中hsdR基因的敲除
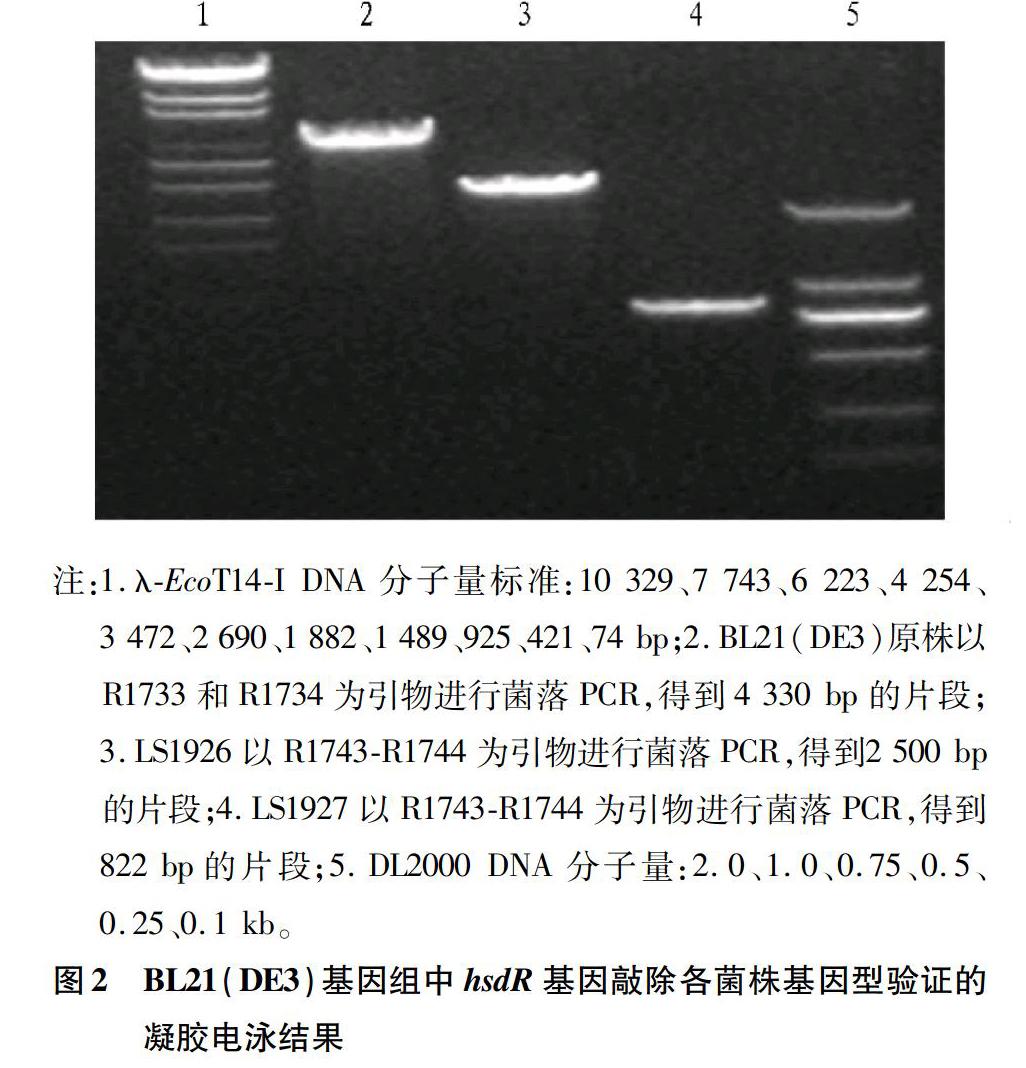
以BL21(DE3)的基因组DNA为模板,分别以R1737和R1738以及R1741和R1742为引物,PCR扩增得到hsdR基因开放阅读框上下游的各500 bp片段;以pLS1662为模板,R1739和R1740为引物,PCR扩增得到1.1 kb的卡那霉素抗性基因。纯化3个PCR产物,合并作为模板,以R1738R1741为引物 PCR扩增得到2.1 kb的融合片段。将此2.1 kb片段电转化至由IPTG诱导表达重组酶的BL21(DE3)/pTKRed电转化感受态细胞,重组酶催化2.1 kb中的2个500 bp片段和基因组中的同源片段进行同源重组,在大观霉素-卡那霉素双抗的筛选作用下,所得的单菌落以R1743和R1744进行菌落PCR来验证抗性变株的基因型,结果如图2所示,获得了与预期大小一致的2.5 kb,表明得到了hsdR基因被两侧含有ISceI作用位点的卡那霉素抗性基因所取代的菌株LS1926,其基因型为BL21(DE3)ΔhsdR∷SneoS,pTKRed。
将LS1926在含100 μg/ml大观霉素的LB培养基中于30 ℃培养,加入1.5% L阿拉伯糖诱导20 h。将培养液稀释106后涂布LB平板,随机挑选150个克隆影印至LB平板和含25 μg/ml卡那霉素的LB平板,所有克隆均表现为卡那霉素敏感性。随机挑取单菌落,以R1743和R1744进行菌落PCR,得到0.8 kb的目的条带(图2)。纯化该片段,以R1743和R1744为引物进行测序,发现与预期完全一致,即卡那霉素抗性基因以及两侧ISceI作用位点1个一段50 bp同源片段得以去除,表明获得了hsdR基因敲除的、无任何冗余片段的变株LS1927,其基因型为BL21(DE3)ΔhsdR,pTKRed。
随后进行pTKRed的消除试验。将所得菌株在无抗LB培养基中于37 ℃培养过夜后,稀释106后涂布LB平板,随后置于42 ℃培养箱进行高温处理。随机挑取25个克隆进行抗性验证,发现其中20个均失去了大观霉素抗性,表明pTKRed得以消除,将此菌株命名为LS1928,其基因型为BL21(DE3)ΔhsdR。
2.2 BL21(DE3)和LS1928电转化效率的比较
为验证BL21(DE3)及其hsdR基因敲除变株LS1928的电转化效率,分别将50 ng的pLS182质粒DNA转化这2个菌株,在含30 μg/ml卡那霉素的LB固体平板上进行筛选,计算总的抗性菌落数。以1 μg的质粒DNA来计算转化效率,即转化效率 = 菌落数/μg DNA。4次平行试验发现,hsdR基因敲除变株LS1928的平均转化效率为4.5×107/μg,而BL21(DE3)原株的平均转化效率为3.1×106/μg,即变株的转化效率提高了14.5倍。
2.3 BL21(DE3)和LS1928质粒酶切的比较
为验证转化质粒的正确性,自含pLS182的克隆宿主菌DH10B以及表達宿主菌BL21(DE3)及其hsdR基因敲除变株LS1928分别提取pLS182质粒进行酶切验证。由图3可见,不同菌株来源所提取的质粒酶切后的带型一致,表明LS1928变株有着与原株一致的质粒扩增环境,hsdR基因敲除不产生影响。
2.4 BL21(DE3)和LS1928蛋白表達的比较
为验证pLS182可在LS1928中表达,分别将pLS182转化到BL21(DE3)和LS1928中。图4为BL21(DE3)原株和LS1928分别表达醛缩酶的结果。由图4可见,2个菌株中蛋白表达情形一致,表明hsdR基因敲除变株LS1928有着与原株BL21(DE3)相同的蛋白表达环境,hsdR基因敲除无影响。
综上所述,hsdR基因敲除变株LS1928的转化效率较BL21(DE3)提高14.5倍,而质粒酶切和蛋白表达的试验证明LS1928的其他功能与BL21(DE3)保持一致,表明了LS1928的适用性。LS1928已保存在中国微生物菌种保藏管理委员会普通微生物中心,保藏编号为CGMCC No.7123。
3 讨论
Red同源重组系统用于大肠杆菌K12系列菌株(如MG1655、DH10B等)成功的例子很多,其同源序列在30~50 bp就能够达到较高的重组效率。有研究表明Red重组系统介导非K12系大肠杆菌重组时,重组效率相对较低[11-12]。
该研究所用的BL21(DE3)是大肠杆菌B系菌株,试验起始,笔者尝试利用基于pSC101质粒骨架,以L阿拉伯糖诱导表达重组酶的体系(如pKD46)和pSIM5热诱导表达重组酶的体系,并分别设计50和500 bp同源臂进行同源重组,结果表明均不能在BL21(DE3)中有效的使用,最终摸索到该文报道的同时携带有Red重组酶基因和IsceI归巢内切酶基因的pTKRed利用500 bp同源臂成功的将hsdR基因敲除。该质粒上的重组酶基因是由异丙基βD硫代半乳糖苷(IPTG)诱导的受LacI基因所调节pLac启动子所驱动;ISceI基因是由L阿拉伯糖诱导的受araC基因所调节的pBAD启动子所驱动。LacI基因和araC基因的调节系统均为严谨调节系统,即不加诱导剂时,启动子下游的基因几乎不表达,这就保证了重组酶表达的严格时空性,不会过量表达而引起异常重组。另外,BL21(DE3)基因组上不含有ISceI酶切位点,从而保证了ISceI酶切的良好专一性。
从突变株中提取的质粒酶切和蛋白表达试验确证了菌株的高转化效率,也证明了质粒在其中的正确复制和表达。高转化效率突变株将减少一个试验步骤,有利于简便快捷地获得突变体蛋白库。高转化效率的蛋白表达菌株将在蛋白质工程等研究领域有着重要的应用,如连接产物的转化效率一般为质粒的1/100,此时10余倍的转化效率将有助于获得更多的转化克隆,即覆盖更多的突变体,有利于迅速地筛选得到目的突变。作为获得高催化效率的催化用酶等工业化研究中的一个关键材料,高转化效率的菌株LS1928将有着广泛的应用空间,也可能获得良好的经济效益。
参考文献
[1] GENOMICS M W.Not junk after all[J].Science,2003,300:1246-1247.
[2] MINOWA N,AKIYAMA Y,HIRAIWA Y,et al.Synthesis and antibacterial activity of novel neamine derivatives[J].Bioorg Med Chem Lett,2006,16:6351-6354.
[3] ZHANG Y,BUCHHOLZ F,MUYRERS J P,et al.A new logic for DNA engineering using recombination in Escherichia coli[J].Nat Genet,1998,20:123-128.
[4] SHARAN S K,THOMASON LC,KUZNETSOV S G,et al.Recombineering:A homologous recombination-based method of genetic engineering[J].Nat Protoc,2009,4:206-223.
[5] POTEETE A R.What makes the bacteriophage lambda Red system useful for genetic engineering:Molecular mechanism and biological function[J].FEMS Microbiol Lett,2001,201:9-14.
[6] TAKAHASHI N,KOBAYASHI I.Evidence for the double-strand break repair model of bacteriophage lambda recombination[J].Proc Natl Acad Sci USA,1990,87:2790-2794.
[7] MURPHY K C.The lambda Gam protein inhibits RecBCD binding to dsDNA ends[J].J Mol Biol,2007,371:19-24.
[8] FERRI L,GORI A,BIONDI E G,et al.Plasmid electroporation of Sinorhizobium strains:The role of the restriction gene hsdR in type strain Rm1021[J].Plasmid,2010,63:128-135.
[9] MANIATIS T.Molecular cloning:a laboratory manual[M].2nd ed.New York:Cold Spring Harbor Laboratory Press,1989:16-34.
[10] KOLISNYCHENKO V,PLUNKETT G,HERRING C D,et al.Engineering a reduced Escherichia coli genome[J].Genome Res,2002,12:640-647.
[11] FRIEDMAN D I,COURT D L.Bacteriophage lambda:Alive and well and still doing its thing[J].Curr Opin Microbiol,2001,4:201-207.
[12] DERBISE A,LESIC B,DACHEUX D,et al.A rapid and simple method for inactivating chromosomal genes in Yersinia[J].FEMS Immunol Med Microbiol,2003,38:113-116.
