磷酸降解棉纤维制备低聚合度葡聚糖的研究
2015-10-13韩华欣刘志华
韩华欣,杨 芳,肖 丹,刘志华,黎 钢
(河北工业大学化工学院,天津300130)
0 引言
近年来,学者们对纤维素降解的研究主要集中在制备较大聚合度的纤维素链段[1]或是葡萄糖[2]、有机酸[3]、乙醇[4]、五羟甲基糠醛[5]等小分子物质,而对于制备低聚合度葡聚糖的研究很少.葡聚糖作为一种具有生物活性的天然化合物,是一种无毒的高附加值产品[6],且不同聚合度的葡聚糖具有不同的活性[7-9].因此对于降解棉纤维制备不同聚合度葡聚糖的研究是很有必要的.目前,降解棉纤维的方法有很多,其中酸降解方法工艺简单成熟,且产率较高.磷酸中的4个O有较强的电负性,更易与纤维素中的羟基形成强的氢键,使H3PO4有效嵌入到棉纤维中破坏其聚集态结构,从而使纤维素链断裂[10-11].同时,磷酸的酸性适中、腐蚀性小,对棉纤维降解温和,可以较好的获得低聚合度葡聚糖.因此,本实验采用质量分数为85%的浓磷酸降解棉纤维,用GPC对降解产物进行测试,得到平均聚合度为100~140、60~100和7的不同低聚合度的葡聚糖,棉纤维降解前后的分子结构和结晶度分别用FT-IR和XRD进行表征.本实验还对10~60葡聚糖迅速降解为水溶性葡聚糖的动力学进行了研究.
1 实验部分
1.1 低聚合度葡聚糖的制备
1.1.1 为100~140葡聚糖的制备
首先进行预处理:将10 g棉纤维放入盛有100 m L质量分数为85%的浓磷酸的烧杯中,室温下超声3 h以加速其溶解.之后将处理过的棉纤维与磷酸的混合溶液放入55℃的水浴锅中,机械搅拌.分别降解4 h、6 h和8 h后,向降解液中倒入100m L去离子水,出现白色沉淀,抽滤,收集滤饼,并用大量的去离子水洗去滤饼中残留的磷酸,再用丙酮冲洗至粉末状.将该白色粉末置于50℃的真空干燥箱中干燥,得到葡聚糖样品I.
预处理过程同1.1.1节.棉纤维与磷酸的混合溶液在55℃下分别降解10 h、12h和14h后,向降解液中倒入不同体积的乙醇,出现白色沉淀,抽滤,收集滤饼,并用大量的去离子水洗去滤饼中残留的磷酸,再用乙醇冲洗至粉末状.将该白色粉末置于50℃的真空干燥箱中干燥,得到葡聚糖样品II.
预处理过程同1.1.1.棉纤维与磷酸的混合溶液在55℃下降解10 h,向降解液中倒入100m L去离子水,抽滤,收集滤液.对滤液进行浓缩,向浓缩后的滤液(大约150 m L)中倒入300 m L丙酮,抽滤,收集滤饼,用丙酮冲洗至粉末状.将该白色粉末置于50℃的真空干燥箱中干燥,得到葡聚糖样品III.
1.2 葡聚糖分子量分布的测定
1.2.1 葡聚糖样品I和II的分子量分布的测定
将葡聚糖样品I和II溶解于8%LiCl/DMAc中[12],得到的浓度为2mg/m l葡聚糖溶液.在凝胶渗透色谱法(GPC)测试之前,将8%的LiCl/DMAc稀释至0.5%.一方面0.5%的含盐量相较于8%溶液的粘度大大降低,可以加速流动相的流动,使得GPC快速分析;另一方面,较低的含盐量可以降低对凝胶色谱柱的损害,延长使用寿命.以0.5%LiCl/DMAc作为流动相,对葡聚糖样品I和II进行GPC测试,以重均分子量分别为46400、15 700、9 600、6 540、2 920的聚苯乙烯作为标准品对其进行分析.
1.2.2 葡聚糖样品III的分子量分布的测定
将葡聚糖样品III溶解于高纯水中,得到浓度为2 mg/m L葡聚糖溶液.以高纯水作为流动相,对葡聚糖样品III进行GPC测试,以重均分子量分别为5 200、11 600、23 800、48 400的葡聚糖和麦芽糖作为标准品对其进行分析.
1.3 葡聚糖样品表征
采用红外结构Vector-22型傅里叶变换红外光谱仪对葡聚糖样品结构进行表征.将样品真空干燥48h后,与干燥的KBr粉末混合研磨、压片,分析的波数范围为0~4 500 cm1.
采用D/Max-2000型X射线衍射仪对葡聚糖样品的结晶度进行表征.测试条件:辐射Cu-K,电压40 kV,电流40mA,扫描范围5°lt;2lt;80°.
2 结果与讨论
2.1 不同的低聚合度葡聚糖的制备条件
棉纤维的天然高结晶度结构很难被低浓度的磷酸破坏,而质量分数为85%的浓磷酸可嵌入到纤维素中破坏其结晶结构.虽然高温有利于加快反应速率,但是在过高温度下,纤维素和磷酸的混合溶液容易发生焦糖化反应而变成黑色[13].因此,反应温度应不高于60℃.而低温条件下纤维素降解缓慢,反应时间较长.综上,为避免焦糖化反应和缩短反应时间,选择55℃作为较佳反应温度.在葡聚糖制备过程中,反应时间和沉淀剂是控制葡聚糖 的重要参量.对为60~140的葡聚糖,均可通过控制反应时间及沉淀剂的极性得到.
棉纤维及其降解后的低聚合度葡聚糖分子量分布见图1.由图1可以看出,经磷酸降解后的棉纤维的降低,且其分散指数(PDI)也有所下降,表明低聚合度葡聚糖样品的分子量分布变窄,即葡聚糖的分子量更为集中.
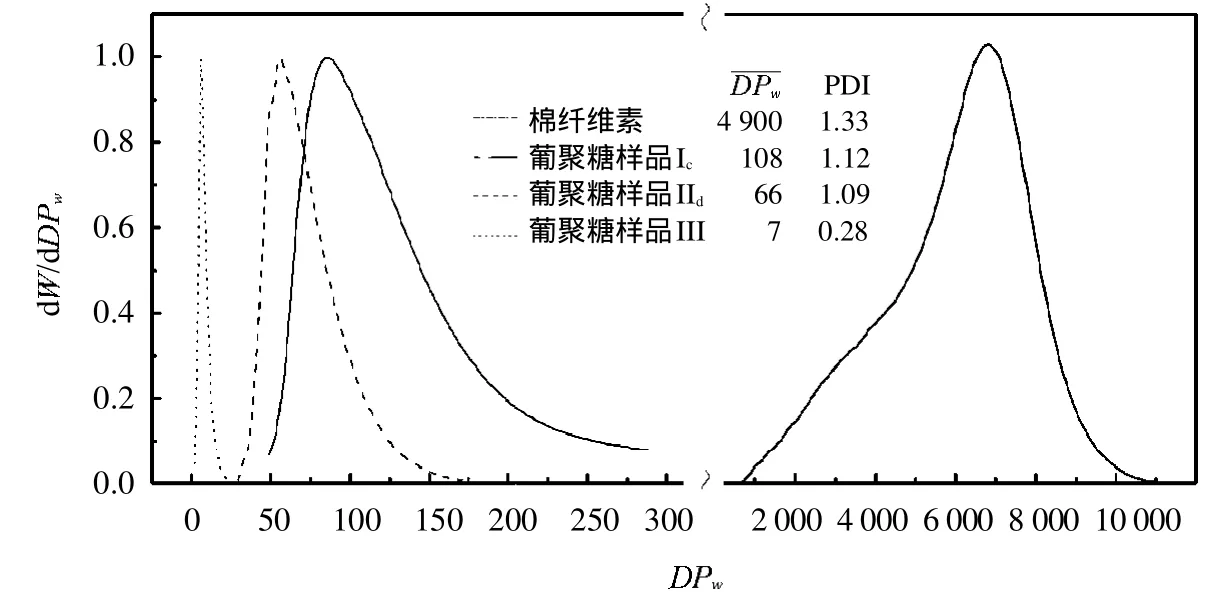
图1 棉纤维素和葡聚糖样品的分子量分布Fig.1 Molecular weight distribution of cotton celluloseand glucan samples
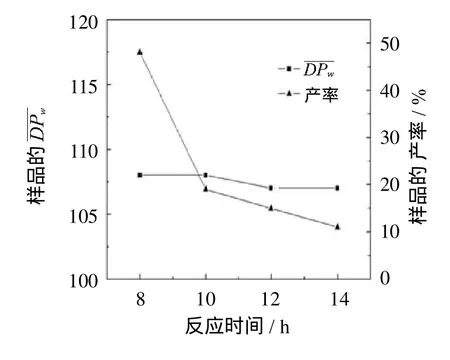
图2 反应时间对葡聚糖样品I的和产率的影响Fig.2 Thee ffect of reaction timeon the and yield of glucan sample I
2.1.2 60~100为的葡聚糖样品II的制备
Liebert[14]通过改变沉淀剂的极性得到了不的水溶性纤维素糊精.由此可见,沉淀剂的极性会对样品有所影响.本实验中将沉淀剂由水改为乙醇.
表1 制备DPw 100~140葡聚糖样品I的优化条件Tab.1 Optim ized operation conditions for preparation of100~140 glucan sample I

表1 制备DPw 100~140葡聚糖样品I的优化条件Tab.1 Optim ized operation conditions for preparation of100~140 glucan sample I
样品编号 反应时间/h 葡聚糖样品产率/% PDI Ia 4 73.14 1.21 131 Ib 6 57.43 1.15 116 Ic 8 48.00 1.12 108
表2 制备DPw 60~100葡聚糖样品II的优化条件Tab.2 Optim ized operation conditions for preparation of60~100 glucan sample II

表2 制备DPw 60~100葡聚糖样品II的优化条件Tab.2 Optim ized operation conditions for preparation of60~100 glucan sample II
样品编号 反应时间/h葡聚糖样品产率/% PDI IIa 10 42.71 1.07 92 IIb 10 49.21 1.11 75 IIc 12 34.36 1.08 85 IId 14 40.86 1.09 66 1 2 1 3
以丙酮作为沉淀剂,对葡聚糖样品I和II的滤液进行处理,得到水溶性葡聚糖样品III.表3为不同反应时间对葡聚糖样品III聚合度和产率的影响.由表3可以看出,随反应时间的延长,样品基本维持在6~7之间,且PDI也基本不发生明显变化.样品产率在反应时间为10h时达到最大,之后开始降低.这是因为相比于大分子量的纤维素,小分子量的水溶性葡聚糖的降解速率更快[17].延长反应时间,小分子量的葡
由以上实验结果可知,在实验中没有制得10~60的低聚糖.这可能是因为该范围内的低聚合度葡聚糖迅速发生降解,生成了水溶性葡聚糖.为此,本研究建立了一个动力学方程来描述水溶性葡聚糖的生成.水溶性葡聚糖生成的动力学模型如式 (1)
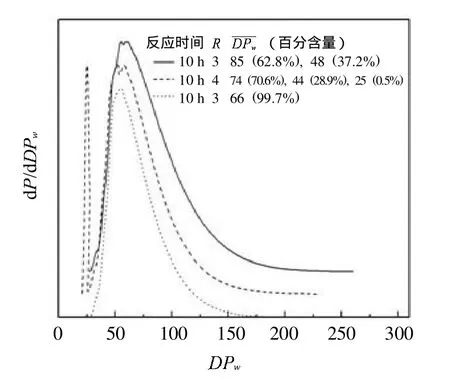
图3 反应时间和R对葡聚糖样品II的 影响Fig.3 Theeffectof reaction timeand R on thef glucan sample II
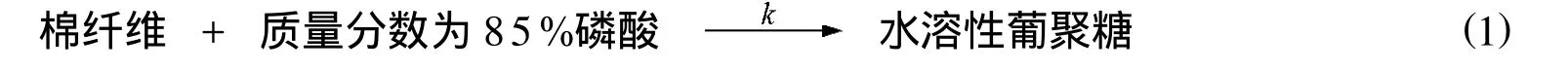
其中:k表示水溶性葡聚糖的生成速率常数.基于动力学模型式得水溶性葡聚糖的生成动力学方程,如式(2)

其中: 为水溶性葡聚糖的浓度,g/m L; 为反应级数; 为反应时间,h.
一般情况下,反应动力学方程是联系浓度(c)和反应时间(t)的方程.在50~60℃范围内,反应级数分别为0、1、2、3时,考察了反应动力学参数的线性关系.结果见表4.
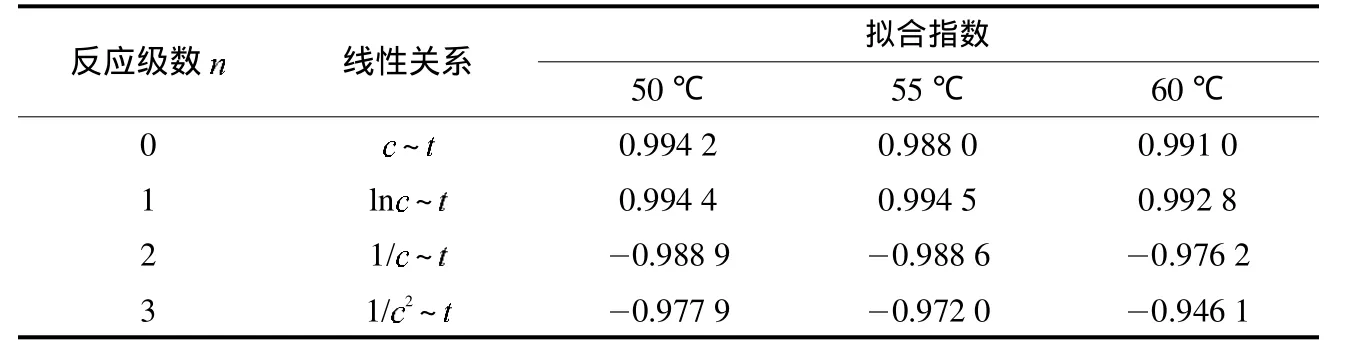
表4 水溶性葡聚糖生成反应动力学的反应级数Tab.4 The reaction orderof the water-soluble glucans generation
通过比较表4中的拟合指数可知,n为1时拟合指数最高.因此,水溶性葡聚糖生成的动力学反应属于一级反应.该结果和Zhang[18]所研究的微晶纤维素的去结晶动力学类似.因此式 (2)可写成

对式 (3)进行积分,得到方程

其中:ci为常数,g/m L,表示棉纤维素中所含水溶性葡聚糖的初始浓度.根据式 (4),结合实验数据以lnc和t做图,如图4所示.图4中的斜率即为速率常数(k ).在50℃、55℃和60℃下,水溶性葡聚糖的生成速率常数分别为0.075h1,0.100h1和0.125h1.该速率常数表明水溶性葡聚糖的生成较快,即表明10~60范围的葡聚糖发生迅速降解,生成了水溶性葡聚糖.其活化能的计聚糖会降解成纤维二糖、葡萄糖和糠醛等小分子产物.
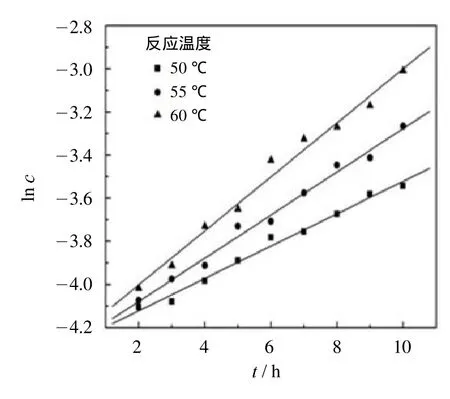
图4 水溶性葡聚糖的生成速率模型Fig.4 Generation ratema thematical modeling of water-solubleglucans
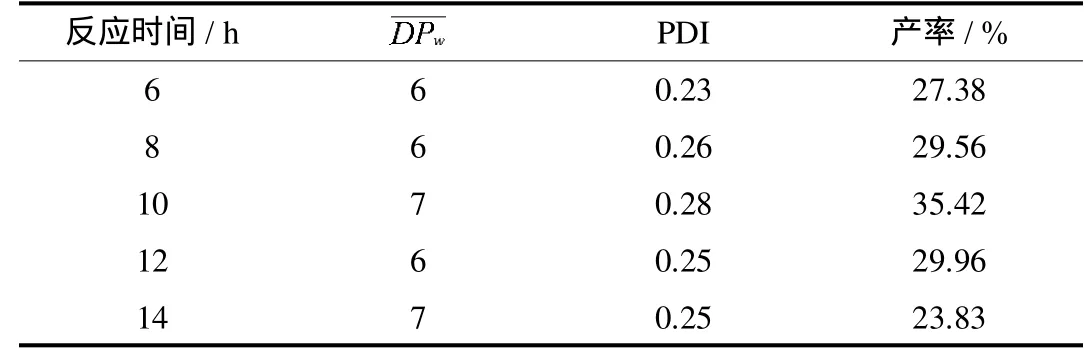
表3 反应时间对葡聚糖样品III的影响Tab.3 The effectof reaction time on glucan sample III
2.2 水溶性葡聚糖生成的动力学研究
算参照阿伦尼乌斯方程[19].

选取50℃和60℃下的速率常数0.075 h-1和0.125 h1,根据式 ( 5)计算在50~60℃的水溶性葡聚糖生成的活化能(Ea)为45.72 kJmol-1,指前因子(A)为1.898×106h-1.廖艳芳等[20]曾指出纤维素热裂解的活化能在低温段为267 kJmol-1,在高温段为174 kJmol-1;Qi[21]曾指出棉短绒溶解于NaOH/尿素体系的活化能为101 kJmol-1.通过对比可知,浓磷酸降解棉纤维的过程中活化能较低,继而导致了范围内的葡聚糖发生迅速降解,生成了水溶性葡聚糖.
2.3 结果表征
2.3.1 红外结构表征
图5为葡聚糖样品III和棉纤维素的红外谱图.由图5可知,原纤维素和葡聚糖样品的红外光谱图差别不大.在3413 cm-1左右,峰形较宽,是纤维素分子内的O-H因氢键缔合而形成[22].由于纤维素分子中含有水分,并且在真空干燥中不能完全除去,所以在1 642 cm-1处出现由H-O-H引起的吸收峰[23].在2 900 cm-1、1 375 cm-1、1 158 cm-1、1 031 cm-1、893 cm-1、665 cm-1的强吸收峰,分别归属于C6位的-CH2的伸缩振动、C-H的变形振动、C-O-C的伸缩振动、C-C和C-O的伸缩振动、 -糖苷键的C-1-H的伸缩振动、平面外C-OH的伸缩振动.从图5可看出没有任何新的吸收峰产生,即在棉纤维素降解过程中,在纤维素链中没有新官能团产生.综上,磷酸在降解棉纤维素的过程中并未对其化学结构造成破坏.
葡聚糖样品III的降解程度远远大于葡聚糖样品I和II,因此,图5的红外谱图分析也同样适用于葡聚糖样品I和II.
2.3.2 结晶度表征
图6为棉纤维素、葡聚糖样品Ic和III的XRD衍射图.由图6可以明显看出,棉纤维素在2 为14.88°、18.80°、22.33°和33.55°处有强衍射峰.对比棉纤维素和葡聚糖样品的衍射图谱,在葡聚糖样品的衍射图谱中,原属于棉纤维素的2 为14.88°和18.80°的结晶峰消失了,而出现了2 为12.20°、19.80°、38.20°和40.70°的新的非结晶峰.这表明了再生纤维素的产生,即棉纤维素的结晶结构已被破坏,结晶度得到降低.图6中,葡聚糖样品衍射图谱在22.33°附近的结晶面积明显降低,而在19.80°处的非结晶峰面积增加,这再次证明了葡聚糖样品中结晶度已得到降低.该实验结果同Deng[24]的研究结果类似.
结晶指数 (Xc)可根据公式 (6)进行计算[25].

其中:Acr和Aam分别表示结晶峰面积(2θ=22.33°处的衍射峰)和非结晶面积(2θ=19.80°处的衍射峰).
根据式(6)得到棉纤维素的结晶度指数为90.6%,葡聚糖样品I和III的结晶度指数分别为47.2%和41.9%.对比结晶度指数发现,葡聚糖样品的结晶度明显下降.
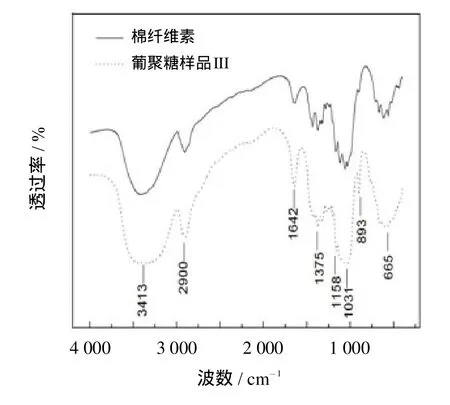
图5 葡聚糖样品III和棉纤维素的红外谱图比较Fig.5 Comparisonsof FT-IR spectraon glucan sample IIIand cotton cellulose
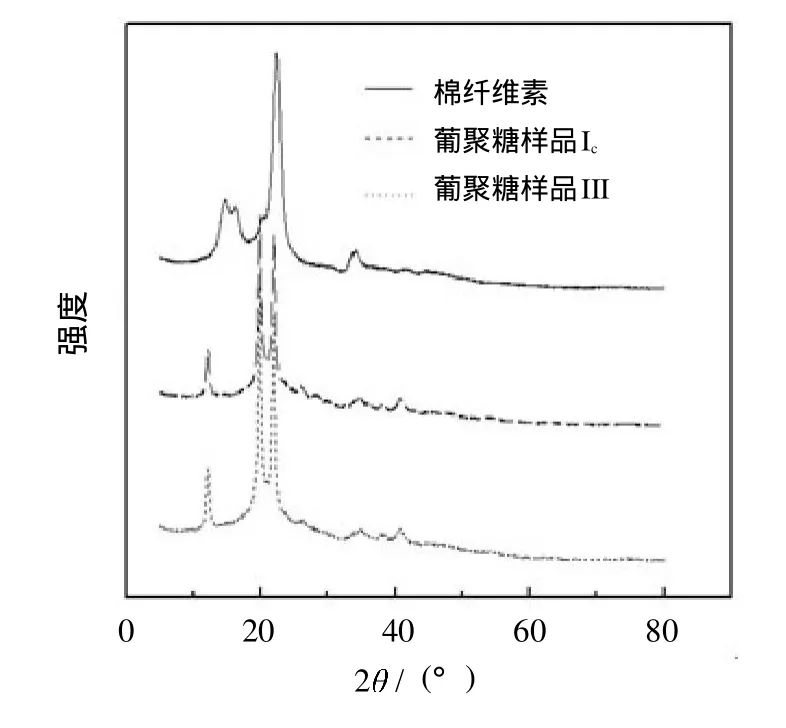
图6 棉纤维素、葡聚糖样品Ic和III的XRD衍射图Fig.6 XRD patternsof cotton celluloseand glucan samples Ic and III
葡聚糖样品II和I均属于水不溶性的葡聚糖,且其 居于样品I和III之间.因此,图6中衍射峰的分析结果同样适用于葡聚糖样品II.
3 结论
本文主要采用了磷酸降解棉纤维素的方法来制备低聚合度葡聚糖.通过控制反应时间和沉淀剂得到了0~140、1007的葡聚糖样品.在降解前后,葡聚糖样品的 和结晶度均得到了有效降低,但其化学结构并未遭到破坏.10~60范围内的葡聚糖迅速降解为水溶性葡聚糖,其反应动力学符合一级反应,在50℃、55℃和60℃下的速率常数分别为0.075 h1、0.100 h1和0.125 h1,其活化能为45.72 kJmol1.
[1]Yu JL,Yang F,Liu ZH,etal.Preparation and characterization of C10-C14alkylcelluloseestersulfatesurfactant[J].JSurfactDeterg,2014,17(4):647-653.
[2]冯建萍,刘民,贾松岩,等.吡咯烷酮酸性离子液体高效催化纤维素水解制葡萄糖 [J].石油学报(石油加工),2012,28(5):775-782.
[3]Yan L,Qi X.Degradation of cellulose to organic acids in itshomogeneous alkaline aqueous solution[J].ACSSustainable Chem Eng,2014,2(4):897-901.
[4]赵鹏翔,赵正凯.磷酸预浸蒸汽爆破玉米秸秆生产纤维素乙醇 [J].酿酒科技,2013(10):34-37.
[5]庞斐,吕惠生,张敏华.亚临界水/二氧化碳中纤维素降解制备5-羟甲基糠醛的机理及动力学[J].化学反应工程与工艺,2007,23(1):55-60.
[6]Vetvicka V.Glucan-immunosti mulant,adjuvant,potentialdrug[J].World JClin Oncol,2011,2(2):115-119.
[7]LiX,Deng F,HuaY,etal.Effectofmolecularweightofdextranon thephasebehaviorandm icrostructureofpreheated soy protein/dextranm ixtures[J].Carbohyd Polym,2008,72(1):160-168.
[8]王双慧,沈南辉,何勇,等.燕麦-葡聚糖对高胆固醇小鼠血脂和游离脂肪酸的影响研究 [J].食品工业科技,2014,35(2):324-327.
[9]郝和群,姚萍.葡聚糖分子量和接枝度对阿霉素/白蛋白-葡聚糖纳米粒子体外抗肿瘤效果的影[J].高等学校化学学报,2014,35(3):652-659.
[10]Kang P,QinW,Zheng ZM,etal.The oretical study on the mechanisms of cellulose dissolution and precipitation in the phosphoric acid-acetone process[J].Carbohyd Polym,2012,90(4):1771-1778.
[11]Fadeeva J,Shmukler L,Safonova L.Investigation of the phosphoric acid-N,N-dimethylformam ide system as potentialsolvent for cellulose[J].JMol Liq,2003,103:339-347.
[12]DupontA L.Cellulose in lithium chloride/N,N-dimethyl-acetam ide,optim isation of adissolutionmethod using paper substratesand stability of the solutions[J].Polymer,2003,44(15):4117-4126.
[13]Jiang B,Liu Y,BhandariB,etal.Impactof caramelization on theglass transition temperature of severalcaramelized sugars.Part I:Chem ical analyses[J].JAgric Food Chem,2008,56(13):5138-5147.
[14]LiebertT,SeifertM,Heinze T.Efficientmethod for thepreparationofpure,water-solublecellodextrines[C]//MacromolSymposia.Wiley-VCH Verlag Gmbh and Co.KGaA,Weinheim,2008,262(1):140-149.
[15]CalviniP,GorassiniA,MerlaniA L.On thekineticsof cellulosedegradation:lookingbeyond thepseudozeroorder rateequation[J].Cellulose,2008,15(2):193-203.
[16]Håkansson H,Ahlgren P,Germgård U.The degree of disorder in hardwood kraft pulps studied bymeans of LODP[J].Cellulose,2005,12(3):327-335.
[17]MosierNS,Ladisch CM,LadischM R.Characterizationofacid catalytic domains forcellulosehydrolysisand glucosedegradation[J].Biotechno Bioeng,2002,79(6):610-618.
[18]Zhang J,Zhang J,Lin L,etal.Dissolutionofm icrocrystallinecellulose in phosphoricacid-molecularchangesand kinetics[J].Molecules,2009,14(12):5027-5041.
[19]Eken-Sara鐎?o lu N,Mutlu SF,DilmaçG,etal.A comparativekinetic study of acidic hem icellulosehydrolysis in corn cob and sunflower seed hull[J].Bioresource Technology,1998,65(1):29-33.
[20]廖艳芬,王树荣.纤维素热袭解过程动力学的试验分析研究 [J].浙江大学学报(工学版),2002,36(2):172-176.
[21]QiH,Chang C,Zhang L.Effectsof temperatureandmolecularweighton dissolution of cellulose in NaOH/ureaaqueoussolution[J].Cellulose,2008,15(6):779-787.
[22]杨芳,黎钢,楚彦芳,等.羧甲基纤维素-丙烯酰胺接枝共聚反应研究 [J].河北工业大学学报,2004,33(5):52-55.
[23]Sun X F,Sun R C,Tomkinson J,et al.Degradation of wheat straw lignin and hemicellulosic polymers by a totally chlorine-freemethod[J].Polym Degrad Stabil,2004,83(1):47-57.
[24]DengW,Tan X,FangW,etal.Conversion of cellulose into sorbitolover carbon nanotube-supported ruthenium catalyst[J].Catal Lett,2009,133(1-2):167-174.
[25]Focher B,PalmaM T,CanettiM,etal.Structuraldifferencesbetween non-wood plantcelluloses:evidence from solid state NMR,vibrational spectroscopy and X-ray diffractometry[J].Ind Crops Products,2001,13(3):193-208.
