稀杯盔形珊瑚转录组分析
2015-09-21刘金豆黄元佳
刘金豆,刘 丽,黄元佳
(广东海洋大学水产学院//南海水产经济动物增养殖广东省普通高校重点实验室//南海生物资源开发与利用协同创新中心 广东 湛江 524088)
稀杯盔形珊瑚转录组分析
刘金豆,刘丽,黄元佳
(广东海洋大学水产学院//南海水产经济动物增养殖广东省普通高校重点实验室//南海生物资源开发与利用协同创新中心 广东 湛江 524088)
以高通量测序技术Illumina HiSeqTM2000对稀杯盔形珊瑚(Galaxea astreata)进行转录组测序分析,共获得50 360 620条短序列 (reads)。利用Trinity软件对所有reads从头组装后得到81 014条单基因簇(Unigenes),60 471条(74.58%)编码蛋白框(Coding Sequences,CDs)。与N(rNon-redundant, 非冗余)、COG(Cluster of Orthologous Groups of proteins, 蛋白相邻类的聚簇)、KEGG(Kyoto encyclopedia of genes and genomes, 京都基因与基因组百科全书)、Swissprot四大数据库比对共获得36 545条注释基因。其中与COG数据库比对获得14 491条注释基因,分为24个功能类别,其中参与一般功能预测类的Unigene数最多,有4 642条;KEGG分析比对获得16 021条注释基因,分成241类,包括代谢通路、钙离子信号通路、MAPK信号通路等,在所有通路中参与代谢途径的基因数最多,共有2 450条(15.29%);GO(Gene Ontology, 基因本体)功能分类将Unigene分为47个类别。
转录组;稀杯盔形珊瑚;Unigene;信号通路
珊瑚礁生态系统是全球最大的生态系统之一,在热带海洋环境中扮演着极为重要的角色。近年由于温度变化、海洋酸化、海水污染等环境因素的影响,导致世界范围内珊瑚礁大面积白化和死亡[1-3]。李淑等[4]在珊瑚礁白化研究进展中从细胞机制和光抑制机制两个方面综述了珊瑚白化的机制,但尚未研究清楚。基因层面上,由于珊瑚研究起步晚,缺乏珊瑚基因信息,使得分子层面上珊瑚的白化机制也十分模糊[5-6]。通过转录组测序技术获得珊瑚基因信息和功能,了解珊瑚的转录组特征,将有助在基因水平上揭示珊瑚的白化机制。
转录组(transcriptome)是指细胞或组织所表达的所有RNA总和。它反映特定条件下细胞或组织内的基因表达情况和生物学代谢途径的调节。转录组测序技术是最近发展起来的利用深度测序进行转录组分析的技术,该技术能提供全面的转录组信息,且具有灵敏度高、分辨率高、重复性好的特性。目前,该技术已经大规模应用。2009年Feng等[7]对静水椎实螺(Lymnaea stagnalis)中枢神经系统进行了转录组分析,获得7 712条EST序列,注释得到了很多基因。同年,Eli Meyer[8]等人首次将高通量测序技术应用到珊瑚上,获得了大量的珊瑚基因和注释信息。2011年Wei等[9]利用该技术进行芝麻转录组研究,并对芝麻转录组整体特征进行了初步的分析,获得86 222条Unigene序列,在很大程度上丰富了芝麻生物信息数据。除此之外,还有大量生物进行了转录组测序分析,如模式生物中的玉米(Zea mays L.)[10-11]、拟南芥(Arabidopsis thaliana (L.) Heynh)[12-13]等;非模式生物中的油菜(Brassica campestris L.)[14]、白鲑(Salmonidae)[15-16]、大比目鱼(Hippoglossus stenolepis)[17]等。
稀杯盔形珊瑚(Galaxea astreata) 隶属于枇杷珊瑚科,盔形珊瑚属。本研究以广东徐闻县珊瑚礁国家级自然保护区一种造礁石珊瑚——稀杯盔形珊瑚为研究对象,进行转录组测序及数据分析。由于受到环境变化的影响,导致保护区内珊瑚礁数量逐年减少,稀杯盔形珊瑚即为受害者之一。自2009年以来,本课题组从分子分类[18-19]、性腺发育[20]以及功能基因克隆[21]等方面对稀杯盔形珊瑚进行了研究。本研究通过高通量测序技术对该珊瑚进行转录组测序,以期丰富该珊瑚的转录组特征和信息,以及为珊瑚礁的保护和修复提供理论依据。
1 材料和方法
1.1材料
稀杯盔形珊瑚(Galaxea astreata )样本均采自广东省湛江市徐闻珊瑚礁国家级自然保护区(20°10′36″N—20°27′00″N)。于室内循环水族箱暂养15 d左右,自然光照11~12 h·d–1, 海水水温为(27±1.0),℃ 每周更换1/3 体积海水并清洗水循环系统。
1.2方法
1.2.1RNA的提取剪取珊瑚虫体3~5个,采用Connolly et al[22]的Trizol法提取珊瑚总RNA, -80℃长久保存。部分RNA送广州基迪奥生物科技有限公司进行检测、建库、组装、测序等。
1.2.2RNA的质量检测和文库的构建稀杯盔形珊瑚总RNA经Aglilent 2100检测,符合转录组RNA 检测标准[质量浓度≥200 ng/μL,总量≥10~15 μg,OD260/280在1.8~2.2范围,RNA Ratio (28S∶18S) ≥1 , RIN≥7.5]后,进行建库。
样品总RNA,用带有Oligo(dT)的磁珠富集mRNA。加入fragmentation buffer将mRNA打断成短片段,以mRNA为模板,用六碱基随机引物(random hexamers)合成第一条cDNA链,然后加入缓冲液、dNTPs、RNase H和DNA polymerase I合成第二条cDNA链,在经过QiaQuick PCR试剂盒纯化并加EB缓冲液洗脱之后做末端修复, 加poly(A)并连接测序接头,再用琼脂糖凝胶电泳进行片段大小选择,进行PCR扩增,测序文库建好后用Illumina HiSeqTM2000进行测序。
1.3数据处理
1.3.1初始数据处理测序产生的原始图像数据经base calling转化为序列数据(raw reads),过滤后得到clean reads。
1.3.2序列组装使用短 reads 组装软件 Trinity做转录组从头组装。首先将具有一定长度overlap的reads连成更长的片段,从这些通过reads overlap关系得到的不含N的组装片段得到组装出来的 Unigene。
1.3.3功能注释及编码蛋白框(CDS)的预测通过blastx将Unigene序列比对到Nr (Non-redundant,非冗余)、COG(Cluster of Orthologous Groups of proteins,蛋白相邻类的聚簇)、KEGG(Kyoto encyclopedia of genes and genomes,京都基因与基因组百科全书)、Swissprot四大蛋白质据库(evalue<0.000 01)。获取跟给定 Unigene 具有最高序列相似性的蛋白,从而得到该 Unigene 的蛋白功能注释信息。对blast比对结果中rank最高的蛋白确定该Unigene的编码区序列,然后根据标准密码子表将编码区序列翻译成氨基酸序列,从而得到该Unigene编码区的核酸序列(序列方向5'->3')和氨基酸序列。最后,与以上蛋白库均比对不上的Unigene用软件ESTScan预测其编码区,获取其编码区的核酸序列(序列方向5'->3')和氨基酸序列。
2 结果与分析
测序共获得50 360 620条clean reads,Q20(质量不低于20的碱基比例)达到96.44%,不确定的碱基比例为0,GC%值为49.65%。由此可见此次转录组测序结果较好,可以进行后续的数据组装和分析。组装拼接后获得81014条unigene,N50(将所有 Unigene 从长到短排序,并依次累加长度。当累加片段长度达到总片段长度的50%时,对应片段的长度和数量,即为Unigene N50长度和数量)为1 096。这些Unigene中长度最长的为18 230 nt,最短的为201 nt,平均长度为751.55 nt。整体看来,本研究测序结果较好。
2.1Unigene长度分布
从稀杯盔形珊瑚的Unigene长度分布(图1)中看出,所有Unigene的长度都大于等于200 nt。长度在200~299 nt的Unigene数目最多为19 869条,其次为长度大于或等于3 000 nt的Unigene数为1 419条。而长度在2 900~2999 nt的Unigene数目最少为149条。
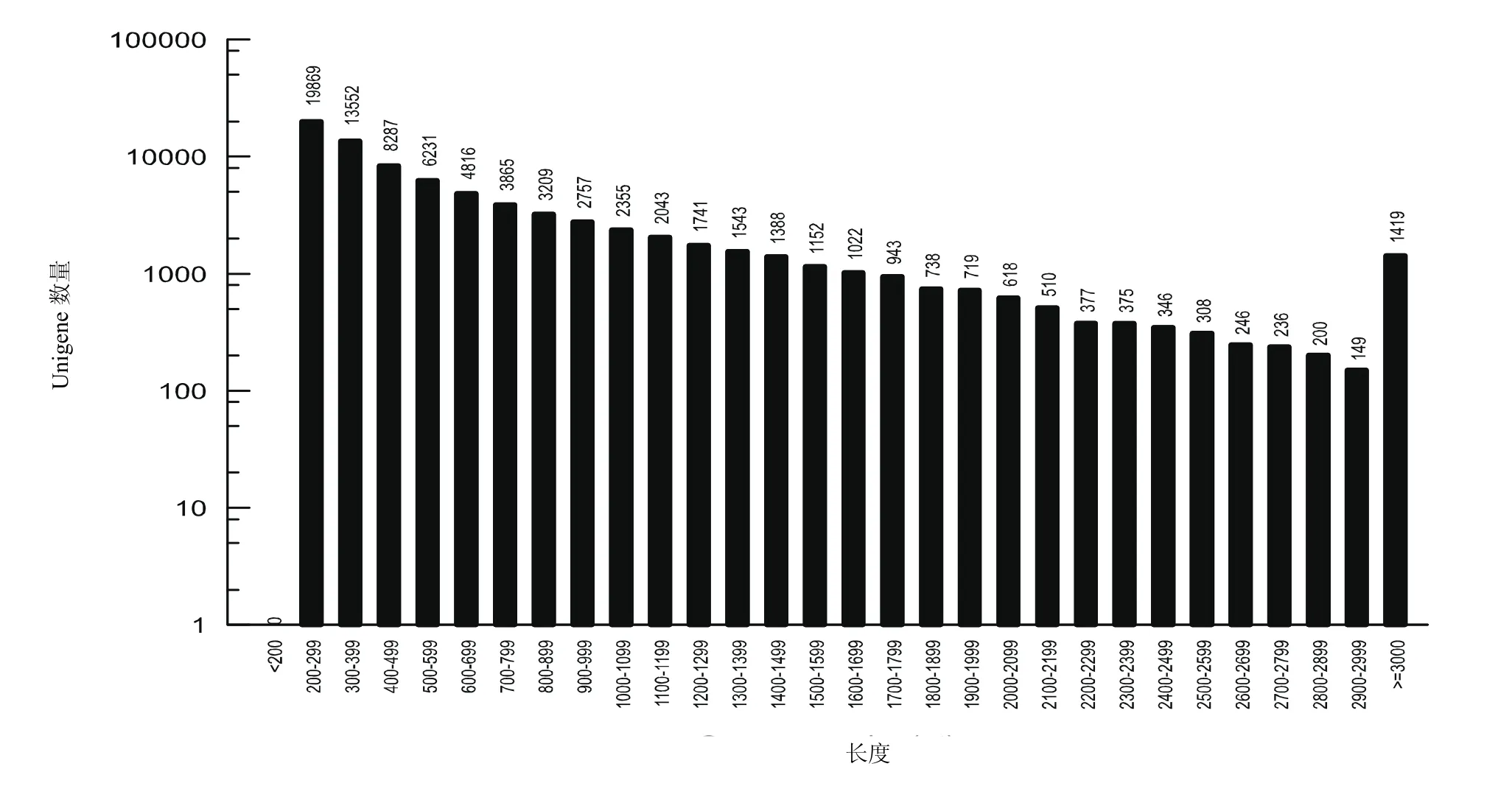
图1 稀杯盔形珊瑚的Unigene长度分布Fig.1 Length distribution of Galaxea astreata-Unigene
2.2Unigene的功能注释
通过与Nr、COG、KEGG、Swissprot等四大数据库进行比对共获得36 545条注释基因。Nr数据库比对注释获得的Unigene数最多,为35 548条。占总Unigene数的43.88%。KEGG数据库比对注释的Unigene数最少,为16 021条,占总Unigene数的19.78%。COG、Swissprot数据库比对注释的Unigene数分别为14 491条和27 872条。未注释的Unigene数为44 469条,占总Unigene的比率为54.89%,超过Unigene总数的一半以上。在Unigene与四大数据库比对注释时,有一些Unigene基因会在数据库中重复出现,多次被注释。稀杯盔形珊瑚的四大数据库注释维恩图简明清晰的展示出四大数据库注释的Unigene之间的关系(图2)。由图2可见,共同注释的Unigene数为9 089条,占注释的Unigene总数的24.87%。

图2 稀杯盔形珊瑚的四大数据库注释维恩Fig.2 Venn of the four databases Galaxea astreata-unigene
2.3Unigene的功能分类
2.3.1Unigene的COG分类COG功能分类将Unigene分为24个类别,如图3所示。从图中看出Unigene的COG功能种类比较全面,几乎包含了所有的生命活动。参与一般功能预测类的Unigene数最多,有4 642条,所占比率为32.03%。细胞核结构功能类别含有的Unigene只有9条,所占比率为0.62%,是所有类别中Unigene数最少的。其他的功能分类详见图3。

图3 稀杯盔形珊瑚的Unigene COG功能分类Fig.3 COG Function Classification of Galaxea astreata-Unigene
2.3.2Unigene的KEGG分类与KEGG数据库进行比对,获得16 021条注释Unigene,分为241条途径,包括代谢通路、MAPK信号通路、钙离子信号通路、蛋白质在内质网加工过程、细胞周期、RNA转运等多种途径。部分途径列于下表1。从表1中可见,代谢途径所占有的Unigene数最多,为2 450,占15.29%。其次为MAPK信号通路,共有Unigene数为657条,所占比率为4.1%。钙离子信号通路所占有的Unigene数为610条,占3.81%,位居第三。

表1 稀杯盔形珊瑚Unigene的KEGG功能分类Table 1 KEGG function Classification of Galaxea astreata-Unigene
2.3.3Unigene的GO分类如图4所示,GO功能分类将注释的Unigene分为3类:生物过程、细胞组成和分子功能,共47个分支。其中生物过程包含23个不同的类别,是三大类别中所含类别最多的一类。所含类别如下:细胞生理过程、新陈代谢的过程、生物调节、生长、死亡等。细胞生理过程包含的Unigene数目最多为8 185条,其次为新陈代谢的过程,有7 192条Unigene。死亡类别的Unigene数最少,只有8条。
细胞组成和分子功能又分别分为13和11个类别。细胞组成中所含Unigene数最多的类别为细胞,拥有7 993条Unigene。细胞部分包含的Unigene数比细胞类只少两条,位列第二。而突触部分仅仅有35条Unigene,是细胞组成中Unigene数目最少的一类。分子功能中的催化活性所占有的Unigene数最多有7 359条。电子转运活性所占Unigene数最少,只有10条。
2.4编码蛋白框(CDs)的预测
通过blastx比对(evalue < 0.000 01)和ESTScan预测Unigene编码区,得到其编码框。两者共得到编码蛋白框60 471条(74.58%),大部分Unigene的蛋白编码框已经得到预测。其中blast比对得到36 564条CDs,占总Unigene 数的比为45.13%,ESTScan预测得到的CDs为23 907条,占Unigene总数的29.51%。各自得到的CDs长度分布图具体如图5和图6所示,由图可知,所得CDs的长度在200~299 nt之间所占比例最大。
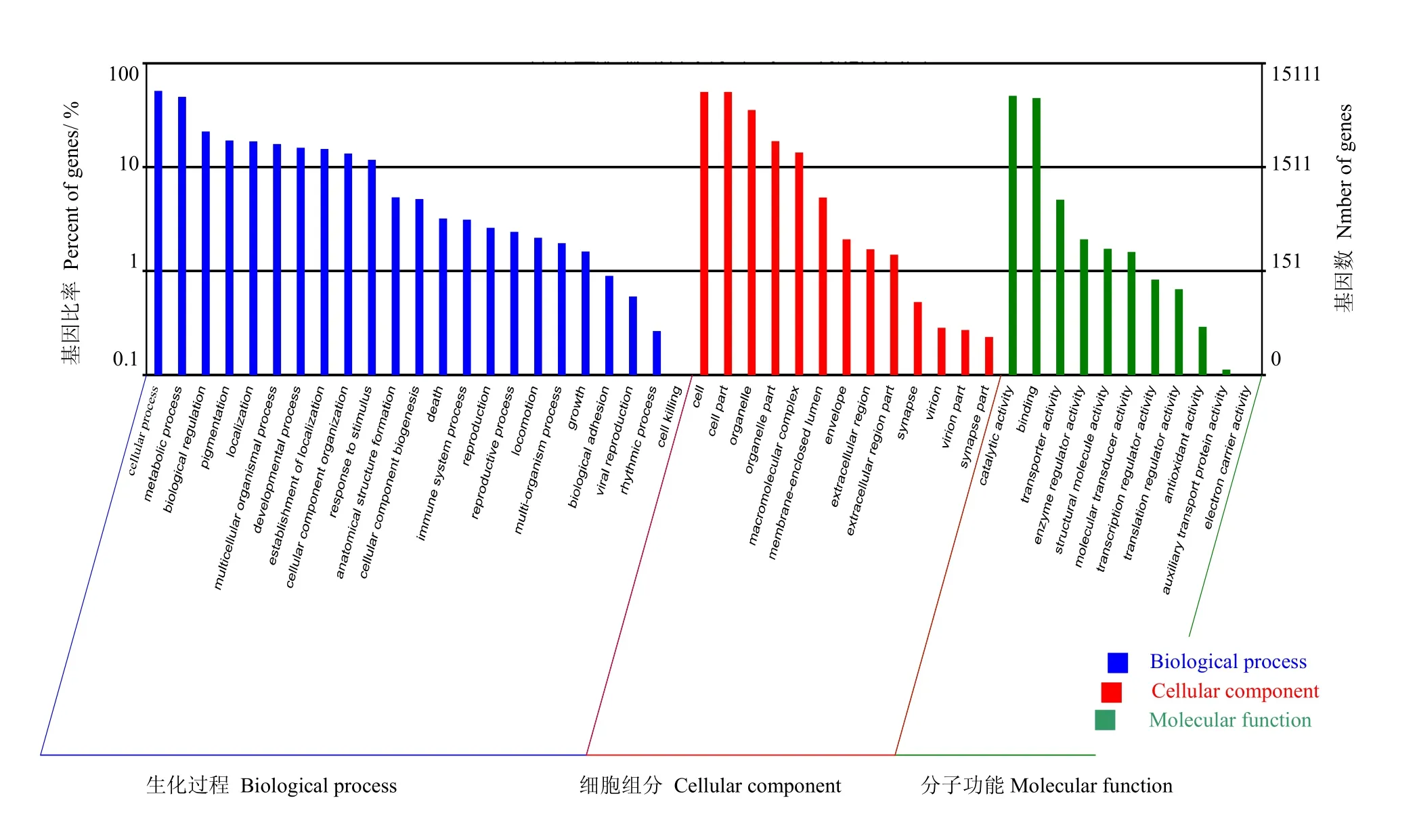
图4 稀杯盔形珊瑚的Unigene GO功能分类Fig.4 GO class of Galaxea astreata-Unigene
3 讨论
本研究通过Illumina HiSeqTM2000对稀杯盔形珊瑚(Galaxea astreata)进行转录组测序,共获得50 360 620 reads,组装后获得81 014条Unigene。比对后得到36 545条注释基因,未注释的基因有44 469条,占总Unigene的百分比为54.89%。出现大量未能注释的Unigene大致包括以下2个因素。
(1)珊瑚基因信息的缺乏。相对于其他物种如:拟南芥、斑马鱼而言,珊瑚的研究起步较晚,目前关于珊瑚基因组和转录组的研究很少,其数据库信息不足,缺乏可供参考的基因组信息。这致使对基因进行功能注释时往往找不到对应的注释信息。
(2)测序技术的局限性。高通量测序技术虽然有很多的优势,但是同时也存在一定的不足。由于珊瑚没有参考基因组信息,只能进行重头转录组测序分析。重头组装测序中序列长度较短的会影响后期数据质量。根据魏利斌等[23]的研究认为转录组序列越短,获得注释信息的可能性就越小。
从实验结果Unigene长度分布图和N50值的大小来看,Unigene长度都是在200 nt及其以上,N50值为1 096(Unigene N50越长,数量越少,说明组装质量越好。),说明本实验获得的数据较好。同时,将NCBI数据库中已有的10条稀杯盔形珊瑚的基因序列与本研究测序结果进blast比对发现,10条序列都能够比对至本实验的数据库中,并且序列之间的相似程度很大,可靠性很高。如,将NCBI中稀杯盔形珊瑚的Fe基因序列(登录号:KJ59 1050.1)与本研究测序的数据库进行本地blast比对,找到了其对应的基因,并且结果显示其S值为1 489 Bits,E值为0.0,表明两序列具有高度同源性,并且可靠性很高,说明本研究测序数据的准确性较高。

图6 ESTscan预测的CDs长度分布Fig.6 Length distribution of ESTscan.CDs
与Nr、COG、KEGG、Swissprot等四大数据库进行比对共获得36 545条注释基因。GO功能分类中细胞生理过程包含的Unigene数目最多为8 185条,其次为新陈代谢的过程,有7 192条Unigene;死亡类别的Unigene数最少,仅有8条。KEGG数据库比对共注释16 021条Unigene,分为241条途径,其中代谢途径的Unigene数最多,有2 450条,所占比例为15.29%。钙离子信号通路的Unigene数为610条,所占比例为3.81%,位居第三。GO功能分类与KEGG Pathway分析结果一致,发现珊瑚代谢相关的Unigene数目几乎都是最多,表明珊瑚在生长生活过程中代谢活动非常旺盛。从KEGG Pathway分类结果看参与珊瑚钙离子信号通路的Unigene数目很多,说明珊瑚钙化在珊瑚生长过程中具有重要作用。Souter P等[24]人的研究表明珊瑚白化中珊瑚Ca2+平衡失调,这说明钙化基因参与调解珊瑚的白化。本研究转录组测序找到了钙离子信号通路,并发现了相关钙化基因,这为研究其调控机制,揭示珊瑚白化机制提供可能,也为珊瑚的保护提供理论基础和研究方向。
广东省徐闻县徐闻珊瑚礁国家级自然保护区给予本研究帮助和支持,特致谢忱!
[1]HARVELL C D,MERKEL S,ROSENBERG E,et al. Coral disease,environmental drivers, and the balance between coral and microbial associates[J]. Oceanography,2007,20(1):172-195.
[2]HOEGH-GULDBERG O,MUMBY P J,HOOTEN A J,et al. Coral reefs under rapid climate change and ocean acidification[J]. Science,2007,318(5857):1737-1742.
[3]HUGHES T P,BAIRD A H,BELLWOOD D R,et al. Climate change,human impacts,and the resilience of coral reefs[J]. Science,2003,301(5635):929-933.
[4]李淑,余克服. 珊瑚礁白化研究进展[J]. 生态学报,2007,27(5):2059-2069.
[5]BANUMS I B. A restoration genetics guide for coral reef conservation[J]. Mol Ecol,2008,17(12):2796-2811.
[6]DAY T,NAGEL L,VAN OPPEN M J,et al. Factors affecting the evolution of bleaching resist- ance in corals[J]. Am Nat,2008,171(2):E72-88.
[7]FENG Z P,ZHANG Z,VAN KESTEREN R E,et al. Transcriptome analysis of the central nervous system of the mollusc Lymnaea stagnalis BMC Genomics[J]. BMC Genomics, 2009,451(10):1471 -2164.
[8]MEYER E,AGLYAMOVA GV,WANG S,et al. Sequencing and de novo analysis of a coral larval transcriptome using 454 GSFlx. BMC Genomics,2009,10(1):219.
[9]WEI W L, QI X Q,WANG L H, et al. Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers[J]. BMC Genomics,2011,12:451.
[10]EMRICH S J,BARBAZUK W B,LI L,et al. Gene discovery and annotation using LCM- 454 transcriptome sequencing[J]. Genome Res,2008,17(1):69–73.
[11]OHTUS K,SMITH M B,EMRICH S J,et al. Global gene expression analysis of the shoot apical meristem of maize(Zea mays L.)[J]. Plant J,2007,52(3):391–404.
[12]JONES-RHOADES M W,BOREVITZ J O,PREUSS D. Genome-wide expression profiling of the Arabidopsis female gametophyte identifies families of small,secreted proteins [J]. PloS Genet,2007,3(10):1848–1861.
[13]WEBER A P M,WEBER K L,CARR K,et al. Sampling the arabidopsis transcriptome with massively parallel pyrosequencing [J]. Plant Physiol,2007,144(1):32-42.
[14]TRICK M,LONG Y,MENG J L,et al. Single nucleotide polymorphism(SNP)discovery in the polyploid Brassica napus using Solexa transcriptome sequencing [J]. Plant Biotechnol J,2009,7(4):334–346.
[15]RENAUT S,NOLTE A W,BERNATCHEZ L. Mining transcriptome sequences towards identifying adaptive single nucleotide polymorphisms in lake whitefish species pairs(Coregonus spp. Salmonidae)[J]. Mol Ecol,2010,19(S1):115–131.
[16]JEUKENS J,RENAUT S,ST-CYR J,et al. The transcriptomics of sympatric dwarf and normal lake whitefish(Coregonus clupeaformis spp,Salmonidae)divergence as revealed by next-generation sequencing[J]. Mol Ecol,2010,19(24):5- 389–5403.
[17]PEREIRO P,BALSEIRO P,ROMERO A,et al. Highthroughput sequence analysis of turbot(Scophthal musmaximus)transcriptome using 454-pyrosequencing for the discovery of antiviral immune genes[J]. PLoS ONE,2012,7(5):e35369.
[18]刘丽,李晓娜,陈育盛,等. 基于线粒体基因的石珊瑚分子系统学研究[J]. 海洋与湖沼,2012,43(4):814-820.
[19]刘丽,陈育盛,李晓娜,等. 基于线粒体Cyt b基因的10种石珊瑚的系统发育关系[J]. 广东海洋大学学报,2011,31 (1):6-11.
[20]金磊. 盾形陀螺珊瑚和稀杯盔形珊瑚性腺发育与生长规律的研究[D]. 湛江:广东海洋大学,2014.
[21]范程辉,刘丽,沈城,等. 稀杯盔形珊瑚铜锌超氧化物歧化酶基因全长cDNA序列的克隆与分析[J]. 热带海洋学报,2015,34(1):83-89.
[22]CONNOLLY M A,CLAUSEN P A,LAZAR J G. Purification of RNA from animal cells using Trizol[J]. Cold Spring Harb Protoc,2006(1): 10-15. DOI:10. 1101/pdb.prot4104.
[23]魏利斌,苗红梅,张海洋. 芝麻发育转录组分析[J]. 中国农业科学,2012,45(7):1246-1 256.
[24]SOUTER P,BAY L K,ANDREAKIS N,et a1. A multilocus, temperature stress-related gene expression profile assay in Acropora millepora,a dominant reef-building coral[J]. Mol Ecol Resour,2011,11(2):328-334.
(责任编辑:陈庄)
Transcriptome Analysis of Galaxea astreata
LIU Jin-Dou,LIU Li,HUANG Yuan-Jia
(Fisheries College,Guangdong Ocean University//Key Laboratory of Aquaculture in South China Sea for Aquatic Economic Animal of Guangdong Higher Education Institutes//South China Sea Bio-Resource Exploitation And Utilization Collaborative Innovation Center,Zhanjiang 524088,China)
High-throughput sequencing technology Illumina HiSeqTM2000 was used to sequence the Galaxea astreata transcriptome,and 50 360 620 reads were got in all.And then assembled software Trinity was used to denovo assembled the reads,getting 81014 Unigenes and 60 471 Coding Sequences(CDs). Blasted with the four database Nr(Non-redundant), COG(Cluster of Orthologous Groups of proteins),KEGG(Kyoto encyclopedia of genes and genomes), Swissprot,36 545 annotated Unigenes were gotten. Blasted with COG database,14 491 unig- enes were gained and according to the function it was divided into 24 categories. 4 642 Unigenes join in the general function prediction only category, which own the most numbers of Unigenes. Blasted with KEGG pathway,16 021 Unigenes were annotated, and it was divided into 241 categories according to different pathways,including metabolic pathway, calcium signaling pathway, and MAPK signaling pathway and so on. Of all the pathways, the number of genes join in the metabolic pathways was maximum in 2450 (15.29%). GO(Gene Ontology)functional classification divided all Unigenes into 47 categories. Through this study,a lot of Galaxea astreata coral Unigenes were gotten. It can help us find some new functional genes and to know about the growth of coraland adaptation to the environment changes.
transcriptome;Galaxea astreata;Unigene;signal pathway
S917.4
A
1673-9159(2015)06-0001-08
10.3969/j.issn.1673-9159.2015.06.001
2015-09-21
国家海洋公益性行业科研专项(201105012);广东省自然科学基金项目(S2011010000269); 广东省海洋渔业科技推广专项(A201308E02)
刘金豆(1989—),男,硕士研究生,研究方向为海洋经济动物发育生物学的研究。E-mail:735677865@qq.com
刘丽,女,教授,主要从事海洋经济动物发育生物学的研究。E-mail:zjouliuli@163.com.
