CaSb2O6∶Bi3+,Eu3+荧光粉的制备和发光性质
2015-09-15陈东菊林利添曹丽伟孟建新
陈东菊 汤 利 林利添 邓 超 曹丽伟 孟建新
(暨南大学化学系,广州 510632)
白光LED,由于其节能、环保以及长寿命等特点,已成为21世纪的新一代光源[1-2]。目前,在用于白光LED的荧光粉中[3-5],蓝色荧光粉和绿色荧光粉都有较高的发光效率,而红色荧光粉的效率较低,其研究相对滞后[6-8]。开发能被紫外转换型芯片有效激发的低成本、窄带发射红光的单一基质荧光粉,是当前LED用荧光粉研究的关键。
碱性金属锑酸盐基质的发光材料[9],如M2Sb2O7(M=Ca,Sr),NaSbO3和 CaSb2O6, 近几年越来越受到人们的关注。CaSb2O6属于六方晶系,空间群为P31m。晶体在结合过程中,Sb5+(d10)离子与周围的6个O原子结合形成[Sb2O6]2-畸变的八面体结构[10],其与Ca2+离子交替堆积,构成层状结构。这些结构属性使得基质CaSb2O6不仅有利于电荷分离[11],而且有利于能量转移[12],因而有利于发光性能的增强。最近报道掺杂Bi3+的CaSb2O6∶Bi3+可以成为一种很好的蓝光荧光粉[13]。而Bi3+离子在很多荧光粉,既可以作为激活离子又可以作为敏化离子,受到人们的重视,如Bi3+掺杂的Gd2O3中Bi3+可作为激活离子发出特征的蓝绿光,同时又可敏化共掺杂的Nd3+的发光[14];Bi3+掺杂的Y3Al5O12中Bi3+可作为激活离子发出特征的紫光,同时又可敏化共掺的Dy3+的发光[15];Bi3+掺杂的YVO4中Bi3+可作为激活离子发出特征的绿光,同时又可敏化共掺的Eu3+的发光[16]。目前已有许多关于Bi3+和Eu3+等稀土离子之间发生能量传递的报道[17-18]。Eu3+离子由于其独特4f6壳层结构,可以产生色纯度较高的红光发射,因此Eu3+经常作为红色荧光粉的激活剂[14,18]。本文在此基础上采用共沉淀法合成 CaSb2O6∶Bi3+,Eu3+荧光粉, 对 Bi3+,Eu3+共掺CaSb2O6∶Bi3+,Eu3+荧光粉的结构、发光性质和发光机理进行了详细的研究。
1 实验部分
采用共沉淀法制备了 Ca1-1.5(x+y)Sb2O6∶x Bi3+,y Eu3+(x、y 分别为掺杂离子物质的量分数,x=0~0.01,y=0~0.2)荧光粉。 分别称取 CaCl2、SbCl3和 Bi(NO3)3,加水溶解,配制成 1.0 mol·L-1CaCl2、1.0 mol·L-1SbCl3和0.01 mol·L-1Bi(NO3)3溶液备用。0.1 mol·L-1Eu(NO3)3溶液由Eu2O3(99.99%)溶于适量硝酸制得。按照化学计量比移取上述 Bi(NO3)3、Eu(NO3)3、CaCl2和 SbCl3溶液,搅拌混合形成澄清溶液A。准确称取0.704 g NaOH 与 0.147 4 g Na2C2O4, 加入 16 mL 水溶解,得到澄清溶液B。将溶液A在搅拌下逐滴加入溶液B中,得到白色沉淀。经静置1 h、离心、水洗、烘干,得到前驱体,再置于900~1 200℃马弗炉中煅烧4 h,冷却后得到样品。实验所用水均为二次去离子水,所用试剂除已标注的外均为分析纯试剂。
采用日本Rigaku公司Dmax-rB型X射线衍射仪(X-ray diffraction,XRD,Cu 靶 Kα 辐射,λ=0.154 06 nm,工作电压为36 kV,工作电流为20 mA,扫描速率为8°·min-1)对样品进行物相分析。采用透射电子显微镜(TEM)(型号:Hitachi-7650)观测样品的颗粒尺寸及形貌。用荧光分光光度计(Hitachi,F-4500)检测样品的荧光。测试样品荧光寿命时使用337 nm N2分子脉冲激光器(Spectra-physics,NL-100)为激发光源,样品的发光信号经透镜聚焦、单色器分光后由光电倍增管(HAMAMATSU 1P28型)探测,高速存储示波器采集荧光衰减曲线数据,再对曲线进行单指数拟合,得到样品的发光寿命。
2 结果与讨论
2.1 CaSb2O6 及CaSb2O6∶Bi3+,Eu3+样品的结构分析
如图1为采用共沉淀法制备前驱体下,在900~1 200℃煅烧4 h所得CaSb2O6样品的X射线粉末衍射(XRD)图。从图中可以看出,在1 100、1 200℃煅烧4 h所得的样品,其衍射峰与标准卡片(JCPDS No.46-1496)符合,表明所得的样品为纯相的CaSb2O6结构。而在900、1 000℃煅烧的样品的XRD图中存在弱的杂峰(如图1中*所示),经分析其杂质可能是Sb2O5。从图中还可以看出,随着煅烧温度的提高,样品的衍射峰越来越尖锐,半峰宽变小。其中1 200℃煅烧4 h所得样品的结晶度更高,且其发光性能也较好,故本实验采用共沉淀法及1 200℃后续煅烧4 h制备样品。文献[19]中报道使用高温固相法制备的CaSb2O6需要在1 200℃煅烧24 h,而本文采用共沉淀法制备的前驱体在1 200℃煅烧4 h就可以获得较好的CaSb2O6。
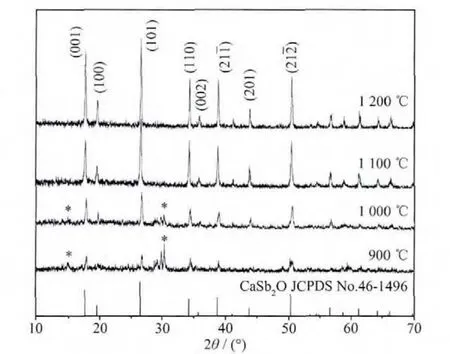
图1 不同煅烧温度所得CaSb2O6的XRD图Fig.1 XRD patterns of CaSb2O6 calcined at different temperature
图 2 为 Ca1-1.5(0.005+y)Sb2O6∶0.005Bi3+,y Eu3+(y=0~0.2)的XRD图。少量掺杂Bi3+、Eu3+离子后,样品的XRD图中没有出现其它的杂峰。当掺杂的Eu3+离子的物质的量分数大于8%时,在2θ=30.33°处出现了可能为Eu2O3的杂峰(如图2中#所示),其他衍射峰的位置都能与标准卡片的衍射峰相对应。Bi3+、Eu3+的离子半径分别为 0.103、0.097 nm,都与 Ca2+离子的半径(0.100 nm)接近, 可以进入 CaSb2O6中的 Ca2+格位[10]。当 Bi3+、Eu3+离子掺杂进入 CaSb2O6,会导致原本的CaSb2O6晶体结构中的键长和对称性等发生变化。 对比未掺杂的 CaSb2O6,Bi3+、Eu3+掺杂的 CaSb2O6的衍射峰位置稍向右偏移,见图3图。虽然共沉淀法使得掺杂的Bi3+、Eu3+离子更容易进入CaSb2O6,但并不能避免晶格空位缺陷的现象。为了保持电荷平衡,基质Bi3+、Eu3+离子掺杂进入CaSb2O6就会产生其它缺陷,比如可能形成[(BiCa)-(VCa)]′、[(EuCa)-(VCa)]′、[(BiCa)-(VCa)-(EuCa)-(BiCa)]·对和 (或)[(EuCa)-(VCa)-(EuCa)][13]簇等结构。
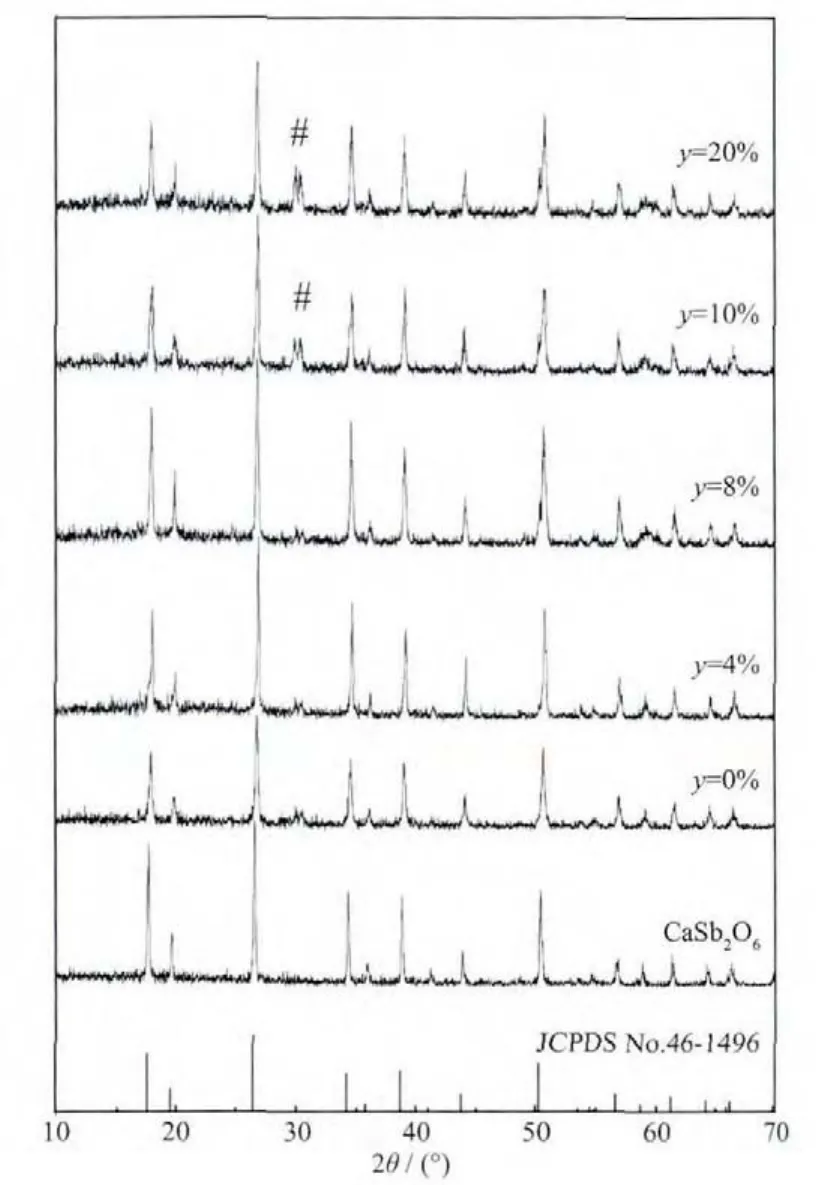
图 2 CaSb2O6 和 Ca1-1.5(0.005+y)Sb2O6∶0.005Bi3+,y Eu3+(y=0~0.2)的 XRD 图Fig.2 XRD patterns of CaSb2O6 and Ca1-1.5(0.005+y)Sb2O6∶0.005Bi3+,y Eu3+(y=0~0.2)

图 3 CaSb2O6、CaSb2O6∶5%Bi3+和 CaSb2O6∶0.5%Bi3+,8%Eu3+的 XRD 图Fig.3 XRD patterns of CaSb2O6,CaSb2O6:5%Bi3+and CaSb2O6:0.5%Bi3+,8%Eu3+
2.2 样品形貌表征
图 4 是 CaSb2O6和 Ca0.8725Sb2O6∶0.5%Bi3+,8%Eu3+的TEM照片。从图中可以看出,所制备样品颗粒均为六边形类圆饼状,尺寸在100~600 nm之间,Bi3+、Eu3+离子的掺入并没有明显改变CaSb2O6的颗粒尺寸及形貌。

图 4 CaSb2O6(a)和 Ca0.872 5Sb2O6∶0.5%Bi3+,8%Eu3+(b)的TEM照片Fig.4 TEM images of CaSb2O6(a)and Ca0.872 5Sb2O6∶0.5%Bi3+,8%Eu3+(b)
2.3 样品的发光性质
2.3.1 CaSb2O6∶Bi3+,Eu3+样品的发光性质
如 图 5 为 Ca1-1.5xSb2O6∶x Bi3+(x=0.25% ,0.5% ,0.75%,2%)系列样品的激发(a)和发射光谱(b)。 从图5可以看出,在458 nm监测所得的激发光谱主要由3 个激发带组成。 以 Ca0.9925Sb2O6∶0.5%Bi3+为例,其峰值波长分别为254、291和336 nm,这与文献[13]报道的基本一致。在291 nm处的激发带来自于CaSb2O6基质晶格的激发,在254 nm处弱的激发峰、336 nm处强的激发峰可分别归属于Bi3+离子的1S0→1P1和1S0→3P1的跃迁。随着Bi3+掺杂量增大,在291 nm处基质的激发带强度增强,这表明了基质晶格与Bi3+离子之间存在能量传递,这也很好解释了在336 nm(1S0→3P1)处的Bi3+强激发峰的半峰宽显著增宽约为40 nm的现象;从图5还可以看出,随着Bi3+掺杂浓度的提高,最强激发谱峰发生明显的红移,从334 nm(0.25%Bi3+)移到362 nm(2%Bi3+)。 在336 nm处激发,样品的发射峰也发生红移现象。这可能是由于Bi3+离子的掺入引起基质的晶体结构发生改变的(如图3),从而使得它的激发光谱和发射光谱发生红移。当Bi3+掺杂量为0.5%时,荧光粉的发光强度最大。

图 5 Ca1-1.5x Sb2O6∶x Bi3+(x=0.25%,0.5%,0.75%,2%)的激发 (a)和发射光谱 (b)Fig.5 Excitation(a)and emission(b)spectra of Ca1-1.5x Sb2O6∶x Bi3+(x=0.25%,0.5%,0.75%,2%)
图6 为在580、351 nm监测下得到的Ca0.8725Sb2O6∶0.5%Bi3+,8%Eu3+的激发和发射光谱。样品的最大激发峰位于351 nm,归属于Bi3+(1S0→3P1)离子的电子跃迁。对比图5,可知这是CaSb2O6共掺杂Bi3+、Eu3+后激发峰发生红移所致。在260、289 nm处弱的肩峰,结合图5,可分别归属于Bi3+离子的1S0→1P1和基质 CaSb2O6的跃迁;393,462 nm 的激发峰归属于Eu3+离子的4f-4f跃迁,分别对应于Eu3+离子7F0→5L6,5D2的跃迁。在351 nm激发下,样品Ca0.8725Sb2O6∶0.5%Bi3+,8%Eu3+的发射光谱中有 7 个发射峰,峰值波长分别是 468、580、594、610、618、629和654 nm。其中468 nm的发射峰是Bi3+离子的发射产生的,580、594、610、618、629 和 654 nm 的发射峰分别对应于Eu3+的5D0→7FJ跃迁。

图 6 Ca0.872 5Sb2O6∶0.5%Bi3+,8%Eu3+的激发和发射光谱Fig.6 Excitation and emission spectra of Ca0.872 5Sb2O6∶0.5%Bi3+,8%Eu3+
2.3.2 Bi3+、Eu3+的掺杂量对 CaSb2O6∶Bi3+,Eu3+发光性质的影响
为了进一步研究Bi3+和Eu3+之间的掺杂量及能量传递的关系,制备了不同Bi3+、Eu3+掺杂浓度的样品。如图7所示,在351 nm激发下监测Ca0.88Sb2O6∶8%Eu3+的发光时,其Eu3+的特征发射强度很微弱。随着 Bi3+的掺入,荧光粉在 580,594、610、618、629 nm处属于Eu3+的特征发射强度呈现快速增加的趋势,这表明了Bi3+能够有效敏化Eu3+的发光。在Bi3+的掺杂量小于0.5%时,样品Eu3+的荧光强度随着Bi3+浓度的增大而增强,这可能是由于Bi3+掺杂浓度的提高,基质与Bi3+离子之间能量传递,促使基质中更多的Bi3+吸收紫外光并将能量传递给Eu3+,使Eu3+的发光强度增强;当Bi3+掺杂量超过0.5%时,样品Eu3+的发光强度明显减弱,这可能是由于在Bi3+的高浓度掺杂下,基质与Bi3+离子之间能量传递达到猝灭中心,属于Bi3+的468 nm处的发射峰强度逐渐降低(如图 7内插图),而且可能形成的[(BiCa)-(VCa)]′、[(EuCa)-(VCa)]′、[(BiCa)-(VCa)-(EuCa)-(BiCa)]·对和(或)[(EuCa)-(VCa)-(EuCa)][13]簇等晶格缺陷作用进一步的加大,其激发能之间的长程迁移导致激发能传递不能被Eu3+完全捕获,因此Eu3+获得的激发能减少,发光强度减弱。如图7内插图所示,当Bi3+掺杂浓度为0.5%时,Bi3+敏化 Eu3+的发光作用最强, 其 Bi3+、Eu3+共掺样品在波长580 nm处的荧光强度是单掺Eu3+样品的10倍左右。
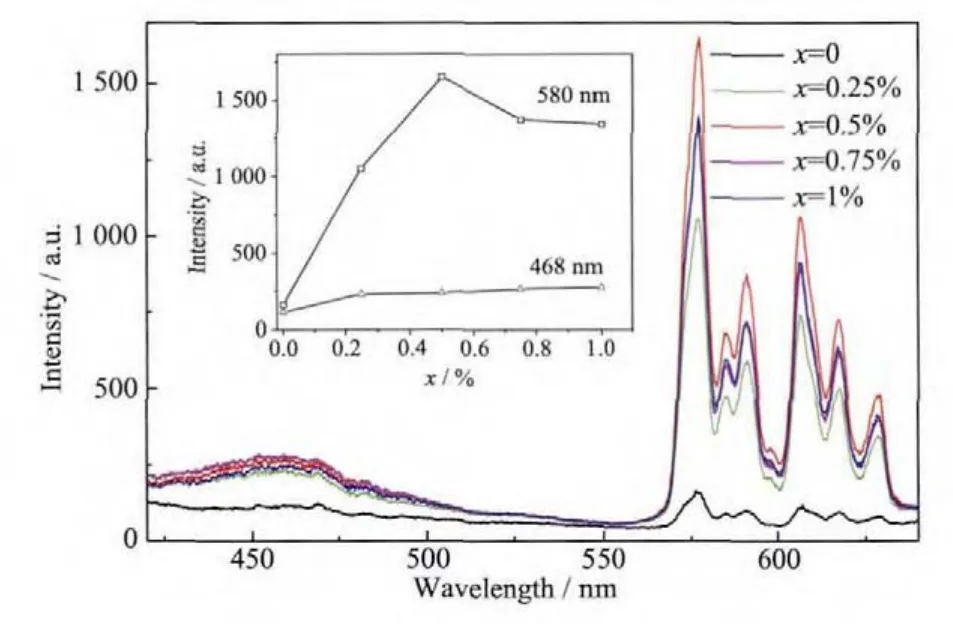
图 7 Ca1-1.5(0.08+x)Sb2O6 ∶8%Eu3+,x Bi3+(x=0~0.01)的发射光谱Fig.7 Emission spectra of Ca1-1.5(0.08+x)Sb2O6∶8%Eu3+,x Bi3+(x=0~0.01)
固定Bi3+的掺杂浓度为0.5%,改变Eu3+的掺杂浓度,在最大激发波 长 351 nm 处激发 Ca1-1.5(0.005+y)Sb2O6∶0.005Bi3+,y Eu3+(y=0.02~0.2)样品时,发射峰的强度随着Eu3+掺杂浓度变化如图8所示。从图8中可知,不同掺杂浓度样品发射光谱的轮廓基本相同,只是强度有变化。如图8内插图,Bi3+离子在468 nm处的荧光强度随着Eu3+掺杂量的增大而单调递减,而Eu3+的特征发射峰大体上随着Eu3+的掺杂浓度增加而增强。这是由于共掺Eu3+后,Bi3+的3P1能级的激发能通过能级跃迁传递到Eu3+的5D0能级,从而导致了Bi3+的荧光强度减弱,Eu3+的荧光强度增强,即在该基质中存在Bi3+→Eu3+的能量传递。属于Eu3+的特征发射峰位于594、610、618、629 nm处的荧光强度大体上随着Eu3+的掺杂浓度增加而增强,这主要是由于增加Eu3+的浓度为基质提供了更多的激活离子,使得更多的Eu3+能够接收来自Bi3+传递的激发能,其发光强度增强。值得一提的是,当Eu3+的掺杂浓度小于8%时,样品Eu3+在580 nm处发光的强度随着Eu3+的浓度增大而明显递增,这可能是由于Eu3+离子占据基质晶格的反演对称中心格位,以磁偶极跃迁为主,样品主要在580 nm处呈橙光(5D0→7F0)发射;而Eu3+的掺杂浓度超过 8%时,其在580 nm处发光的强度稍有减弱,在618和629 nm处发射峰呈现的红光整体上比位于580 nm的 (5D0→7F2)橙光强度更高,这可能是由于随着掺杂离子浓度的不断增大,宇称选择定则会发生松动,导致Eu3+在晶体中处于非反演中心,会以电偶极跃迁为主,样品渐变为橙红光(5D0→7F2)发射的比例更高。

图 8 Ca1-1.5(0.005+y)Sb2O6∶0.005Bi3+,y Eu3+(y=0.02~0.2)的发射光谱Fig.8 Emission spectra of Ca1-1.5(0.005+y)Sb2O6∶0.005Bi3+,y Eu3+(y=0.02~0.2)
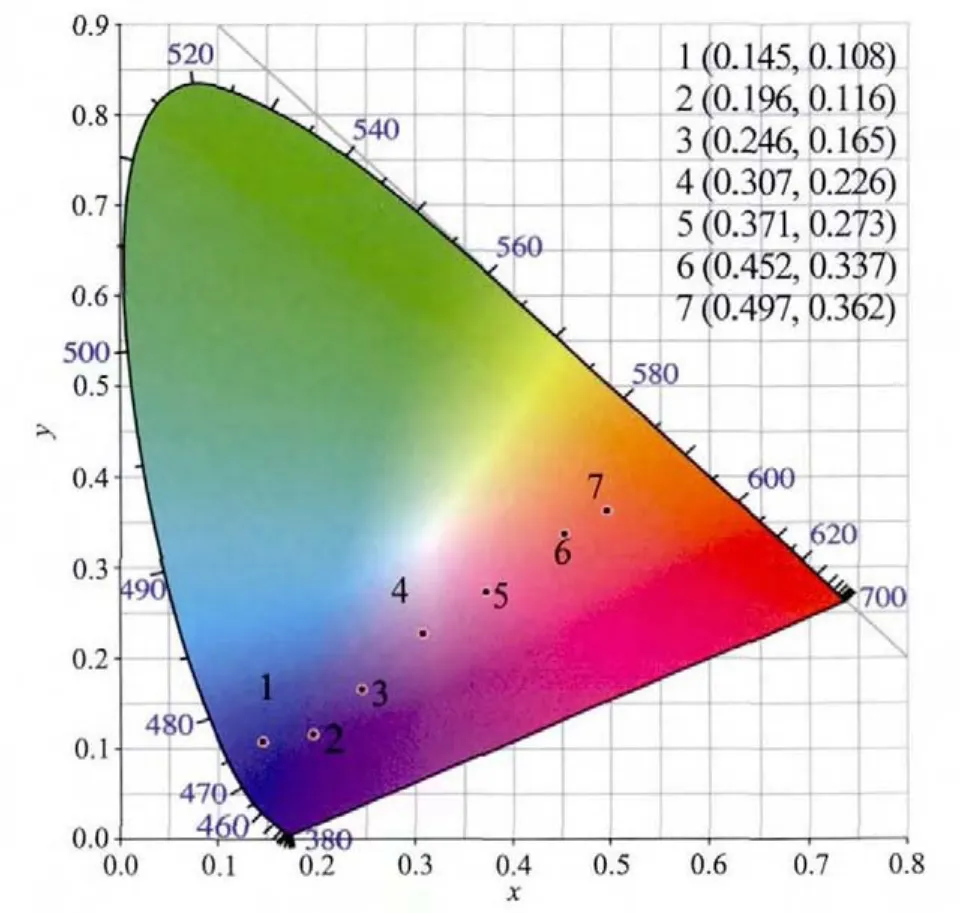
图 9 Ca1-1.5(0.005+y)Sb2O6∶0.005Bi3+,y Eu3+的 CIE1931 色度图 (图中 1~7 分别对应 y=0,0.02,0.04,0.08,0.1,0.15,0.2)Fig.9 CIE1931Chromaticity Coodinates of Ca1-1.5(0.005+y)Sb2O6∶0.005Bi3+,y Eu3+(from No.1 to No.7 corresponded to y=0,0.02,0.04,0.08,0.1,0.15,0.2,respectively)
CaSb2O6∶Eu3+,Bi3+荧光粉的发光颜色和 CIE 色坐标随Eu3+和Bi3+掺杂浓度的变化呈现有规律的变化,如图9所示。当Eu3+的掺杂浓度为0%时,荧光粉呈强的蓝色发射, 其色坐标为 (0.145,0.108);当Eu3+的掺杂浓度为20%时,荧光粉呈强的橙红色发射,主要发射波长分别为580 nm的橙光和610 nm的红光,色坐标为(0.497,0.362)。 当 Eu3+的掺杂浓度在0~8%间变化时,荧光粉的发射颜色从蓝色到红色的过渡;在Eu3+和Bi3+的掺杂浓度分别为8% 和0.5%时,其发射接近白色,色坐标为(0.307,0.226)。因此通过调节Bi3+,Eu3+的掺杂比,该荧光粉可成为一种色坐标可调的荧光粉。
如图10所示的样品在Bi3+的发射波长468 nm处的荧光衰减曲线,进一步证实了荧光粉CaSb2O6∶Bi3+,Eu3+中 Bi3+→Eu3+之间的能量传递。从图 10 可以看出,随着Eu3+掺杂量的增加,Bi3+的荧光衰变速度逐渐加快。对荧光衰减曲线进行单指数拟合后得到荧光寿命 τ*,通过公式 1/τ*=∑kf(kf为衰减速率)[20],ηT=1-kD/kAD(kD为不掺Eu3+时样品的衰减速率;kAD为掺 Eu3+时样品的衰减 速率)[21],可得出样 品 Ca1-1.5(0.005+y)Sb2O6∶0.005Bi3+,y Eu3+(y=0,0.02,0.04,0.08,0.1,0.15,0.2)Bi3+→Eu3+之间的能量传递效率 ηT,结果如图 10内插图所示。可以看出,随着Eu3+的掺杂量的增加,Bi3+→Eu3+能量传递效率逐渐升高,当Eu3+掺杂量为20%时,Bi3+能量传递效率达到了84.8%。
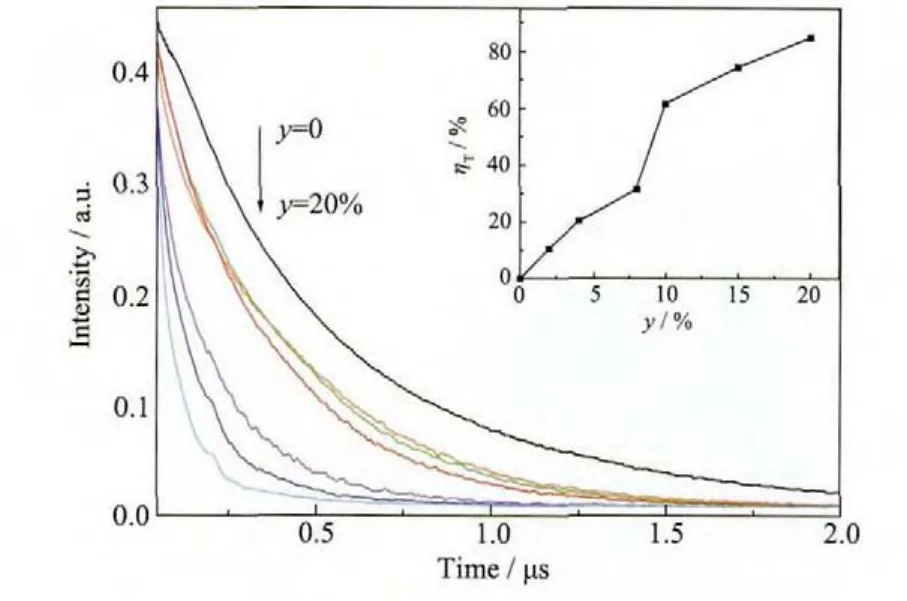
图 10 Ca1-1.5(0.005+y)Sb2O6∶0.005Bi3+,y Eu3+(y=0~0.2)在 Bi3+468 nm发射的荧光寿命衰减曲线Fig.10 Fluorescence decay curves of Bi3+emission monitored at 468 nm for Ca1-1.5(0.005+y)Sb2O6∶0.005Bi3+,y Eu3+(y=0~0.2)
3 结 论
采用共沉淀法及1 200℃后续热处理,成功地制备了荧光粉 CaSb2O6∶Bi3+,Eu3+。 Bi3+对 Eu3+的发光存在高效的敏化与能量传递。该荧光粉能被紫外光/蓝光激发,当Bi3+掺杂浓度为0.5%时,Eu3+掺杂浓度为8%,Eu3+位于580 nm(5D0→7F0)处的荧光发射显著增强,Bi3+,Eu3+共掺样品的荧光强度是 CaSb2O6∶Eu3+的10倍左右。通过调节Bi3+、Eu3+离子掺杂比,荧光粉的发光可呈现从蓝光、白光到红光的变化,表明该荧光粉可实现从蓝、白光到红光的自由调控,可以发展出适用于白光LED用的新型荧光粉。
[1]Narendran N,Maliyagoda N,Bierman A,et al.Light-Emitting Diodes:Research,Manufacturing,and Applications IV:Vol.3938.Yao H W,Ferguson I T,Schubert E F,USA:SPIE,2000:240-248
[2]Nakamura S,Senoh M,Mukai T.Appl.Phys.Lett.,1993,62(19):2390-2392
[3]Li Q,Gao L,Yan DS.Mater.Chem.Phys.,2000,64(1):41-44
[4]Park JK,Lim M,Kim CH,et al.Appl.Phys.Lett.,2003,82(5):683-685
[5]Reineke S,Lindner F,Schwartz G,et al.Nature,2009,459(7244):234-238
[6]Nishida T,Ban T,Kobayashi N.Appl.Phys.Lett.,2003,82(22):3817-3819
[7]Neeraj S,Kijima N,Cheetham A K.Chem.Phys.Lett.,2004,387(1):2-6
[8]Sohn K S,Park D H,Cho SH,et al.Chem.Mater.,2006,18(7):1768-1772
[9]Sato J,Saito N,Nishiyama H,et al.J.Photochem.Photobiol.A,2002,148(1):85-89
[10]Mizoguchi H,Woodward P M.Chem.Mater.,2004,16(25):5233-5248
[11]Singh J,Bhardwaj N,Uma S.Bull.Mater.Sci.,2013,36(2):287-291
[12]Tang JW,Zou Z G,Ye J H.Angew.Chem.Int.Ed.,2004,43(34):4463-4466
[13]Chen L,Long Y,Qin Y,et al.Mater.Lett.,2013,102:59-61[14]Liu G X,Zhang R,Xiao Q L,et al.Opt.Mater.,2011,34(1):313-316
[15]Mu Z,Hu Y,Chen L,et al.J.Lumin.,2011,131(8):1687-1691
[16]Takeshita S,Watanabe T,Isobe T,et al.Opt.Mater.,2011,33(3):323-326
[17]Luwang M N,Ningthoujam R S,Srivastava S K,et al.J.Am.Chem.Soc.,2011,133(9):2998-3004
[18]Chen L,Chen K J,Lin CC,et al.J.Comb.Chem.,2010,12(4):587-594
[19]Lin X,Wu J,Lue X,et al.Phys.Chem.Chem.Phys.,2009,11(43):10047-10052
[20]Lakowicz J R.Principles of Fluorescence Spectroscopy.3rd Ed.New York:Springer,2006.
[21]Vergeer P,Vlugt T J H,Kox M H F,et al.Phys.Rev.B,2005,71(1):014119
