L-去氧肾上腺素合成研究进展
2015-07-24李国华张文博石兆瑞耿佩佩邢荣芬
李国华,张文博,石兆瑞,耿佩佩,邢荣芬
(河北工业大学化工学院,天津 300131)
去氧肾上腺素(phenylephrine,PE)侧链Cα为手性碳原子,其R(-)-异构体的α1-肾上腺素受体 激动作用远强于S(+)-异构体[1-3]。因具有血管收缩作用,PE 的R-构型左旋体在临床上广泛应用于阵发性室上性心动过速,治疗休克及麻醉时维持血压,在眼科检查中可作为安全的短效扩瞳药使用[4-6]。作为伪麻黄碱最简单有效的替代品,去氧肾上腺素在非处方感冒药中的使用也呈现增长趋势[7-8]。
1927年德国化学家Helmut Legerlotz 首次合成了去氧肾上腺素,之后又分离得到其手性异构 体[9-11]。1933年底特律的 Frederick Stearns & Company 公司将其作为减充血剂使用[12]。1976年,美国食品和药物管理局( Food and Drug Administration,简称FDA)将去氧肾上腺素列为缓解感冒、过敏性鼻炎、鼻窦炎导致的鼻塞的安全有效非处方药。随着伪麻黄碱的受限,去氧肾上腺素作为替代品被广泛使用,包括辉瑞、强生等世界各大厂家均在逐步利用去氧肾上腺素来改变其感冒药配方。例如,辉瑞公司将以伪麻黄碱为有效成分的速达菲撤架,推出了以去氧肾上腺素为有效成分的速达菲PE。作为治疗休克与室上性心动过速药物,2010年6 月上海禾丰制药有限公司推出的盐酸去氧肾上腺素注射液获得了中华人民共和国国家食品药品监督管理总局(CFDA)的批准。2014年6 月,美国FDA 批准Éclat Pharmaceuticals L.L.C.公司的新药VazculepTM(盐酸去氧肾上腺素注射剂)上市。去氧肾上腺素越来越受到重视。
目前全球范围内对L-去氧肾上腺素的年需求量约为300t,主要市场为美国、日本、东南亚、欧洲、南美等国家和地区[13-14]。近年来中国与印度取代欧洲Boehringer-Ingelheim 公司成为主要的生产地,其产量分别占全球总产量的50%和40%。在我国获得CFDA 批准的生产厂家主要有深圳沃兰德药业有限公司、赤峰艾克制药科技股份有限公司、潍坊幸福药业有限公司等,且均已形成规模。L-去氧
肾上腺素市场竞争的焦点是产品品质与生产成本,两者都归结于合成路线、生产工艺的选择。目前众多厂家生产路线各异,本文综述了主要的L-去氧肾上腺素合成工艺路线,并将其分为两大类,即拆分法和不对称合成法,而不对称合成法又分为化学不对称合成法和生物不对称合成法。通过工艺对比,对合成方法的优缺点进行了评价,并对未来发展趋势进行了预测。
1 拆分法合成L-去氧肾上腺素
L-去氧肾上腺素作为具有光学活性的药品,传统的合成工艺是合成去氧肾上腺素消旋体,然后通过拆分剂进行手性拆分。下面分别从外消旋体的合成及外消旋体的拆分两方面进行介绍。
1.1 外消旋去氧肾上腺素的合成
外消旋去氧肾上腺素的合成是手性拆分的前提。早在1927年,德国化学家Helmut Legerlotz 就通过钯或铂金属催化剂催化羰基加氢得到外消旋去氧肾上腺素[9]。1951年德国化学家Bergmann 和 Sulzbacher[15]报道了外消旋去氧肾上腺素合成的方法。他以间羟基苯甲醛(1)作为起始原料,β-羟基酸叠氮化物的Curtius 重排反应[16]为关键步骤合成了外消旋去氧肾上腺素(图1)。1961年Russell 和Childress[17]使用同样的起始原料合成了外消旋去氧肾上腺素,是以氢化铝锂作用下的酰胺还原反应为关键步骤(图2)。1974年Hussain 等[18]以间羟基苯乙酮(2)为原料,以10%钯碳催化酮羰基的加氢还原为关键步骤合成了外消旋去氧肾上腺素(图3)。

图1 β-羟基酸叠氮化物Curtius 重排合成路线

图2 氢化铝锂还原酰胺合成路线

图3 钯碳催化羰基还原合成路线
为寻求更加简短、温和的合成路线,2014年Teerawutgulrag 等[19]以间羟基苯甲醛(1)为起始原料,通过wittig 反应[20]构建双键,以双键环氧化胺化(Ⅰ)或双键羟卤化胺化(Ⅱ),得到外消旋去氧肾上腺素,总产率分别为71%和66%。该反应路线步骤简单,各步收率高,避免使用叠氮化合物和氢化铝锂,反应条件温和,有较高的实用价值(图4)。
1.2 外消旋去氧肾上腺素的拆分
1.2.1 先拆分再外消旋化的方法
最广泛使用的L-去氧肾上腺素拆分工艺是利用L-(+)-酒石酸[21]作为拆分剂,生成的D-去氧肾上腺素-L-(+)-酒石酸盐结晶析出,氨水碱化后,经水洗、干燥得D-去氧肾上腺素。而L-去氧肾上腺 素-L-(+)-酒石酸盐留在母液中,加氨水回收得L-去氧肾上腺素[13](图5)。得到的D-去氧肾上腺素在酸性或碱性条件下外消旋化[22],可再次进行拆分,从而提高整体产率。
1.2.2 先拆分再构型翻转的方法
上述拆分工艺,母液得到的L-去氧肾上腺素光学纯度低,需多次精制才能得到合格的L-去氧肾上腺素,并且需在酸或碱的作用下进行催化消旋,总收率低,成本高。李渤等[23]进行了工艺改进,其工艺主要包括拆分和构型翻转[24]两步骤。拆分步骤:首先将DL-去氧肾上腺素、拆分剂D-(-)-酒石酸和异丙醇溶剂混合,依次进行加热反应、降温结晶、固液分离,得L-去氧肾上腺素-D-(-)-酒石酸盐和母液。L-去氧肾上腺素-D-(-)-酒石酸盐加氨水反应,经抽滤、水洗、干燥得L-去氧肾上腺素,母液用于构型翻转;构型翻转步骤:将母液减压蒸馏回收醇溶剂得浓缩液,浓缩液加氨水碱化,得D-去氧肾上腺素,然后依次加甲苯溶剂及乙酸酐,以RCOO-为亲核试剂通过SN2反应得到构型翻转的酯,加80%硫酸,进行水解反应,经氨水碱化得L-去氧肾上腺素(图6)。该工艺使用D-(-)-酒石酸作为拆分剂,比使用L-(+)-酒石酸结晶时间短,一次拆分收率高,并且得到的D-去氧肾上腺素通过构型翻转形成的L-去氧肾上腺素粗品光学纯度>85% ee,与盐酸成盐再碱化即可得到合格的L-去氧肾上腺素,不用再进行拆分,总收率高,成本低。
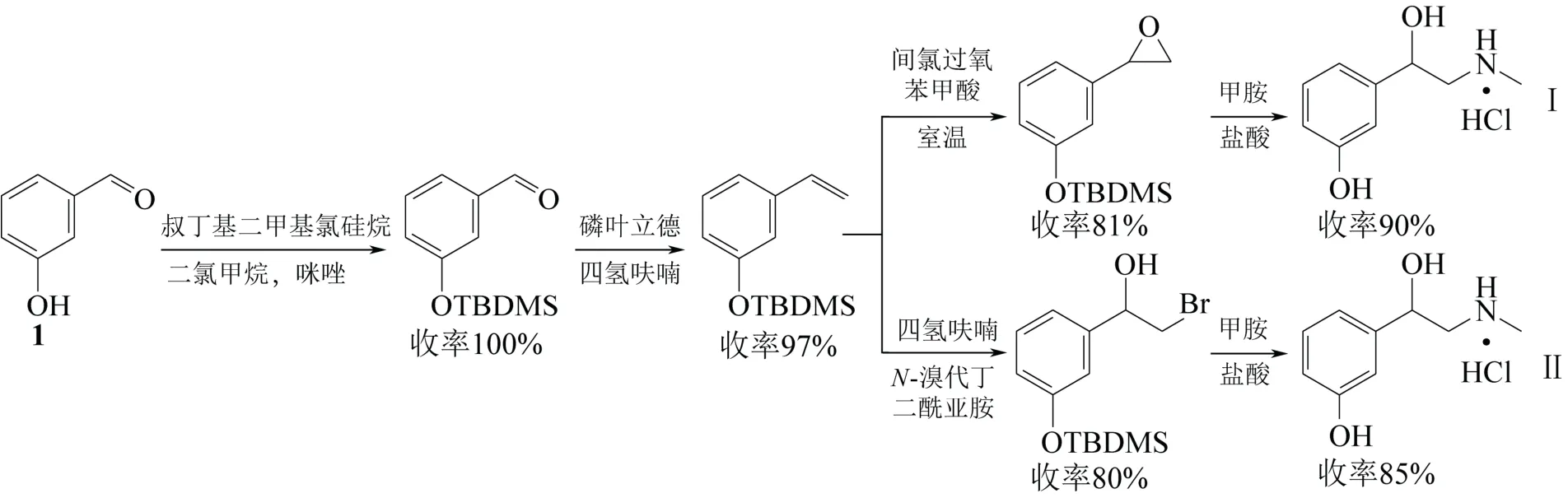
图4 外消旋去氧肾上腺素合成的两种新工艺

图5 拆分再外消旋化的工艺

图6 拆分再构型翻转的工艺
1.2.3 与萘普生拆分工艺串联的方法
Prasad Divi 等[25]发明了一种拆分方法,他们利用L-萘普生[26]作为拆分剂拆分DL-去氧肾上腺素,得到L-去氧肾上腺素和D-去氧肾上腺素,得到的D-去氧肾上腺素进行构型翻转得L-去氧肾上腺素(图7)。这样就可将萘普生与去氧肾上腺素的拆分工艺串联起来,将萘普生拆分后得到的副产品L-萘普生作为拆分剂应用到去氧肾上腺素的拆分工艺中,实现拆分工艺串联,从而提高整体的经济性。1.2.4 水解动力学拆分方法
Gurjar 等[27]通过水解动力学拆分[28]合成了L-去氧肾上腺素。该路线以间羟基苯甲醛(1)为原料,经羟基保护和醛基的环氧化得到外消旋环氧化物,之后再利用手性Salen-Co(Ⅲ)-OAc催化剂[29]对外消旋环氧化合物进行水解动力学拆分,得到光学活性的R-环氧化物和S-二醇。R-环氧化物经甲胺胺化和脱羟基保护得到L-去氧肾上腺素,光学纯度为97% ee(图8)。该方法不仅能得到光学纯度高的产品,并且生成的副产物S-二醇也是重要的医药中间体,然而就合成L-去氧肾上腺素而言,仅此一步就使整体路线的产率低于50%,同时所用的Salen 催化剂价格较贵。
通过上文所述可以看出,拆分法合成L-去氧肾上腺素,先生成外消旋体再拆分,整体路线长。一次拆分理论上可得到50%的产品,剩余的50%虽然可再次进行消旋或构型翻转,但步骤繁杂,各步母液不易处理。得到的L-去氧肾上腺素光学纯度低,需多次精制才能得到合格产品。
动态动力学拆分(DKR)[30]是一种理论产率达100%的拆分新技术,可理解为动力学拆分(KR)[31]与原位消旋反应的耦合。它是在动力学拆分的基础上引入消旋催化剂,使无效的对应体进行原位消旋,实现了拆分过程与底物的消旋在同一体系中相继或同时进行,即某一对映体不断转化为产品,另一无效对映体不断被消旋化,从而使产物的理论产率达100%。目前外消旋仲醇的DKR 研究已受到人们的极大关注,能满足仲醇KR 的催化剂非常多,大多数为脂肪酶,能用于仲醇的DKR 研究的消旋方法主要有碱催化消旋、沸石类酸性催化剂催化消旋以及过渡金属类催化剂催化消旋等。去氧肾上腺素作为手性仲醇类药物,其动态动力学拆分工艺还未见报道,可将动态动力学拆分的新技术应用到L-去氧肾上腺素的拆分工艺中(图9),这对于L-去氧肾上腺素拆分工艺的改造及优化具有重要意义。
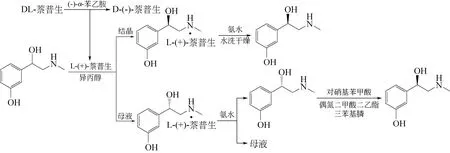
图7 萘普生拆分串联工艺
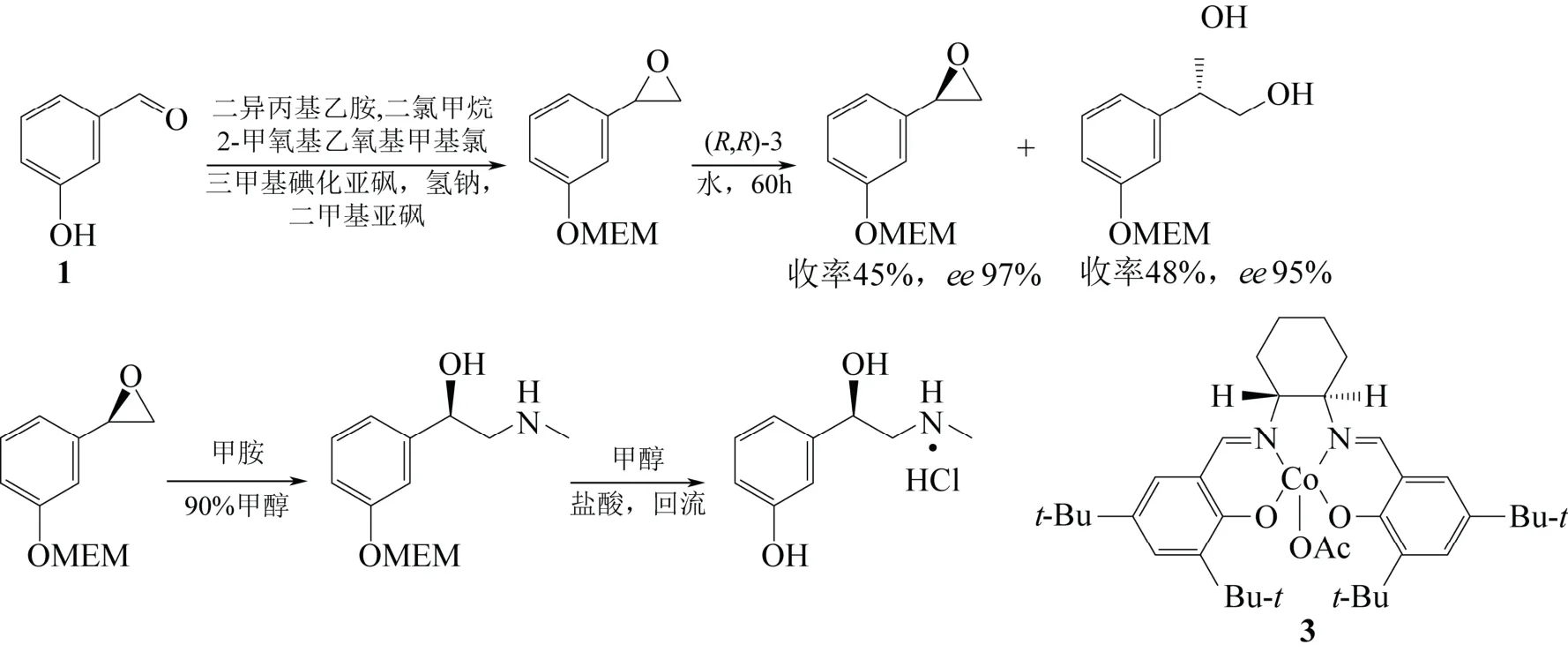
图8 水解动力学拆分工艺

图9 动态动力学拆分工艺
2 不对称合成法合成 L-去氧肾上 腺素
拆分法因步骤繁杂,各步母液不易处理,导致收率低、成本高、污染重、产品光学纯度低等问题。而不对称合成法,因其特有的手性单位增殖效应,目前已成为合成手性药物最经济、有效的方法。不对称合成法可分为化学不对称合成法与生物不对称合成法。化学不对称合成法适于工业大规模生产,生物不对称合成法产品光学纯度高,反应条件温和。
2.1 化学不对称合成法合成L-去氧肾上腺素
在L-去氧肾上腺素的化学不对称合成法中,研究最早、最普遍的方法是不对称催化氢化法。前手性酮的不对称氢化反应是合成具有光学活性仲醇的重要途径。除此之外,Sharpless 不对称双羟基化法、Salen 催化醛的Henry 反应法也是合成L-去氧肾上腺素的有效途径。
2.1.1 不对称催化氢化法合成L-去氧肾上腺素
不对称催化氢化法合成L-去氧肾上腺素是指芳基氨基酮衍生物在手性催化剂作用下氢化还原,生成手性仲醇,再加以处理得到L-去氧肾上腺素。在不对称催化氢化反应中,手性催化剂的结构(手性配体与中心过渡金属)起关键作用。因此人们对于不对称催化氢化合成L-去氧肾上腺素的研究也主要集中在新型手性配体以及中心过渡金属的选择。目前已应用到该体系的过渡金属主要是铑和钌,而手性配体则是以手性双膦配体为主。
铑-手性双膦催化剂催化不对称氢化合成L-去氧肾上腺素,大多是以3-苄基氧基-2-(N-苄基-N-甲基)-氨基苯乙酮盐酸盐(4)为原料,以[Rh(COD)Cl]2为催化剂前体,与不同的手性双膦配体配位形成手性催化剂,催化羰基的不对称氢化反应生成手性仲醇,再经脱苄等步骤得到L-去氧肾上腺素[32]。例如,早在1989年Takeda 等[33]就通过双齿手性双膦配体(2R,4R)-MCCPM(5)与催化剂前体[Rh(COD)Cl]2配位,催化3-苄基氧基-2-(N-苄基-N-甲基)-氨基苯乙酮盐酸盐(4)的不对称氢化反应,经脱苄得到L-去氧肾上腺素,S/C(底物和催化剂的摩尔比)为2000,产品光学纯度为85% ee。Sakuraba 等[34]做了大量研究,发现双齿手性双膦配体(2S,4S)-MCPM(6)和(2S,4S)-MCCPM(7)也具有该功能,S/C为1000,产品ee 值均高达85%。德国BoehringerIngelheim Pharma GmbH & Co.KG 公司[35]对Takeda研究的工艺作出改进,仍然采用(2R,4R)-MCCPM(5)配体与[Rh(COD)Cl]2催化剂前体,只是不对称催化氢化反应后并不立即进行脱苄,而是先将其转换为游离碱,再用氨/甲醇/水的混合溶液沉淀纯化,直接得到99% ee 的产品,之后再进行脱苄,从而减小后期纯化成本,该工艺当S/C 为10000 时仍有很好的选择性。以上实例均见图10。

图10 铑-手性双膦催化剂催化的不对称氢化工艺
Hideo Takeda 和Shunji Sakuraba 虽然利用铑手性双膦催化剂催化合成了L-去氧肾上腺素,但仅能得到85% ee 的产品,需昂贵的纯化成本才能用药,并且使用的催化剂量大,S/C 分别为2000 和1000,不适合工业化。相比而言,德国Boehringer Ingelheim Pharma GmbH & Co. KG 公司发明的工艺在脱苄前,转化为游离碱后通过氨/甲醇/水的混合溶液简单纯化就达到了99% ee 的光学纯度,节省了后期纯化成本,并且S/C 为10000,大大减小了催化剂用量,因此可扩大到工业规模。这些研究中不对称氢化的步骤均是在2MPa 的中压下进行的。
除以上利用铑手性双膦催化剂外,钌系手性双膦催化剂也有很好的效果。铑-手性双膦催化剂因合成路线很长,所用试剂不稳定,合成条件苛刻,价格昂贵。相比而言,钌-手性双膦催化剂制备方法简单,并且对含氧量及含水量的要求并不严格,因此便宜许多。关于钌手性双膦催化不对称氢化合成L-去氧肾上腺素的报道大都以α-甲氨基-间羟基乙酰苯(8)为原料,通过不同的钌手性双膦催化剂催化合成。例如,2008年Hossein[36]最先报道了钌手性双膦催化剂不对称催化氢化合成L-去氧肾上腺素的方法。他们以α-甲氨基-间羟基乙酰苯(8)为原料,形成硫酸盐后,在3MPa 下利用钌-手性双膦催化剂进行不对称催化氢化,之后再用氨水中和得L-去氧肾上腺素,S/C 为3600,反应时间为48h,产品光学纯度为95% ee(图11 Ⅰ)。2010年McGarrity 等[37]则直接用钌双配体催化剂[(R)-Xyl-P-Phos RuCl2(R)-DAIPEN]催化α-甲氨基-间羟基乙酰苯(8)的不对称氢化反应得到L-去氧肾上腺素。(R)-Xyl-P-Phos(9)为C 手性双膦配体,(R)-DAIPEN(10)为手性二胺配体,S/C 为20000,反应时间为16h,产品光学纯度为96% ee(图11 Ⅱ)。2011年冯文化等[38]利用商品化的(R)-BINAP-Ru(II)类催化剂催化合成了L-去氧肾上腺素。(R)-BINAP(11)为C-手性双齿双膦配体,S/C 值为2000,反应时间为45h,还原所得粗品光学纯度为75% ee,经甲醇结晶后可达96% ee(图11 Ⅲ)。
可以看出,Hossein 等的研究相对于McGarrity及冯文化的报道多了一个先酸化再碱化的过程。而冯文化的报道虽然催化剂廉价易得,但还原后只能得到75% ee 的产品,需经甲醇结晶才能得到96% ee的产品。相比而言, McGarrity 的研究不仅可一次还原得到96% ee 的产品,并且S/C 值为20000,反应时间为16h,均优于Hossein 与冯文化的研究。这些研究也都是在3MPa 左右的压力下进行的。
通过上文,可知德国 Boehringer Ingelheim Pharma GmbH & Co. KG 公司发明的铑手性双膦催化工艺和McGarrity 发明的钌手性双膦催化工艺是相对较优良的工艺。然而两者比较而言,McGarrity的工艺省略了脱苄等步骤,合成路线更为简短,所用的钌手性双膦催化剂价格更便宜,催化剂的用量也降低了一倍。
2.1.2 其他化学不对称合成法合成L-去氧肾上腺素 除不对称催化氢化反应外,Sharpless 不对称双羟基化[39]和不对称Henry 反应[40]也成功地应用到L-去氧肾上腺素的合成中。例如,Rajesh Kumar Pandey 等[41]以间羟基苯甲醛(1)为起始原料,以Sharpless 不对称双羟基化反应为关键步骤合成了L-去氧肾上腺素,所得产品的光学纯度为98% ee(图12)。2012年Kureshy 等[42]以间甲氧基苯甲醛(12)为原料,利用Cu(Ⅱ)-大环 Salen 络合物[43]催化醛与硝基甲烷的不对称Henry 反应生成手性硝基醇,之后经三步简便的反应就可得到L-去氧肾上腺素,并有效地对Salen 催化剂进行了回收,产率为85%,产品光学纯度为95% ee(图13)。

图12 Sharpless 不对称双羟基化合成路线

图13 不对称Henry 反应合成路线
以上两种方法均以芳香醛衍生物为原料,成功避开了带压操作,反应温和。相比而言,Sharpless不对称双羟基化法在不对称氧化之前,需要先经过Wittig 反应形成双键,而不对称Henry 反应法不仅可直接通过芳香醛与硝基甲烷的加成反应得到手性醇,并且成功引入了氮原子,使氨基的构建更为方便,同时该反应还实现了Salen 催化剂的回收复用。
2.1.3 路线综合对比
将不对称催化氢化法、Sharpless 双羟基化法、不对称Henry 反应法进行综合对比,如表1 所示。
从表1 中可以看出,以上几种方法中,不对称催化氢化法以芳基氨基酮为原料,后续步骤不用构建氨基,而Sharpless 不对称双羟基化法和不对称Henry 反应法均需在后续步骤构建氨基。从反应类型而言,不对称催化氢化法为还原反应,Sharpless不对称双羟基化法为氧化反应,不对称Henry 反应法为加成反应。就反应条件而言,不对称催化氢化法需2~3MPa 的压力,Sharpless 双羟基化法和不对称Henry 反应法则可有效避免压力反应。这3 种方法均能达到较高的ee 值和产率。从涉及的过渡金属而言,不对称Henry 反应法所需的Cu 比不对称催化氢化法和Sharpless 不对称双羟基化法所需的铑、钌、锇廉价。整体而言,这3 种方法都需要价格较贵的催化剂,大都涉及铑、钌、锇等贵金属。因此开发非贵金属元素催化剂,实现催化剂的高效回收利用是今后研究的主要内容。

表1 化学不对称合成路线对比
2.2 生物不对称合成法合成L-去氧肾上腺素
生物催化具有高选择性、反应条件温和等特点。近年来,不断有生物催化合成L-去氧肾上腺素的报道[44-47]。目前醇氰裂解酶、全细胞酶、酮还原酶、醇脱氢酶等已成功应用到L-去氧肾上腺素的合成工艺中。
LenoX 等[48]提出R-醇氰裂解酶催化的不对称氰醇路线合成L-去氧肾上腺素。他们利用从巴丹苦杏仁提取的R-醇氰裂解酶(PaHNL)为生物催化剂,在pH 值为5.5,温度为0℃的条件下,催化HCN和间甲氧基苯甲醛(12)的加成反应,得到R-手性氰醇,之后通过氰基还原、胺基酰基化、羰基还原、三溴化硼脱甲基等一系列反应,得到L-去氧肾上腺素,产品的光学纯度为99% ee(图14)。
Tokoshima 等[49]以全细胞酶作为生物催化 剂[50],在安伯莱特XAD-7 树脂[51]存在的条件下,选择性还原2-氯-1-(3-硝基苯基)乙酮(14)为(L)-2-氯-1-(3-硝基苯基)乙醇(15)。其中安伯莱特XAD-7树脂可以作为固体“双相有机溶剂”并储存所述底物和产物。该步骤可达99.2% ee 的光学纯度以及87%的分离收率。(L)-2-氯-1-(3-硝基苯基)乙醇(15)再经环氧化、胺化、氨基保护[52]、硝基还原、重氮 化[53]、水解反应可得到L-去氧肾上腺素(图15)。
王波等[54]以商品化酮还原酶KRED 185 作为生物催化剂,以NADP(烟酰胺腺嘌呤二核苷酸磷酸)作为电子供体,催化底物1-(3-羟基苯基)-2-[甲基(苯基甲基)氨基]乙酮(16)为手性醇,再利用无水甲醇和湿钯碳进行脱苄得到L-去氧肾上腺素,光学纯度为99.99% ee(图16)。
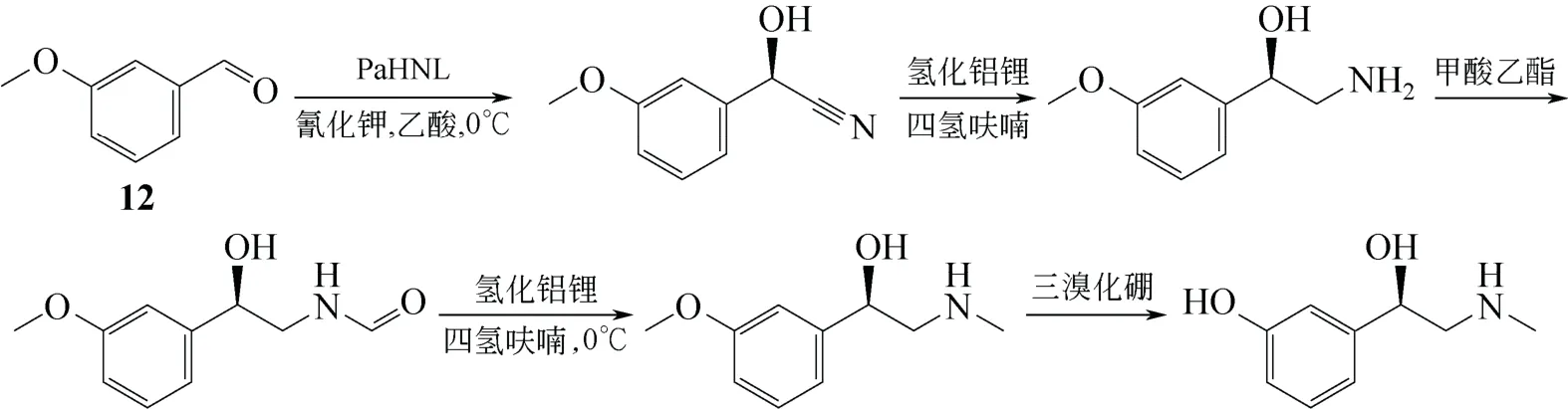
图14 R-醇氰裂解酶催化的不对称氰醇路线
2013年巴斯夫欧洲公司的Breuer 等[55]公开了固氮弧菌属物种EBN1 的醇脱氢酶催化合成L-去氧肾上腺素的方法,该酶可在大肠杆菌中重组制备。该路线是以间羟基苯乙酮(2)为起始原料,丙醇存在的情况下与磺酰氯反应得到α-氯代-3-羟基苯乙酮(17),之后以固氮弧菌属物种EBN1 的醇脱氢酶作为生物催化剂,以NADP 作为电子供体,在葡萄糖脱氢酶和异丙醇存在的条件下,将α-氯代-3-羟基苯乙酮(17)还原成手性醇,甲胺胺化后得到L-去氧肾上腺素,产品光学纯度大于99% ee(图17)。
以上路线对比如表2 所示。
与化学法相比,生物法催化L-去氧肾上腺素的不对称合成,具有很高的选择性,通常ee 值都在99%以上,在王波等[54]利用酮还原酶KRED 185 合成L-去氧肾上腺素的研究中,最终产品ee 值甚至接近100%。此外,这些方法中不对称催化的步骤均是在常压下进行的,反应条件温和。王波报道的酮还原酶催化工艺和Breuer等报道的乙醇脱氢酶催化工艺,均不需要保护基,路线很简短。虽然生物法具有高选择性、反应温和的优势,但也存在着不可避免的问题,如容量低、产率低、反应速度慢、后处理时存在相分离、酶的稳定性和使用寿命受限以及酶的回收再生等问题,还处于实验研究阶段,尚未实现工业化。
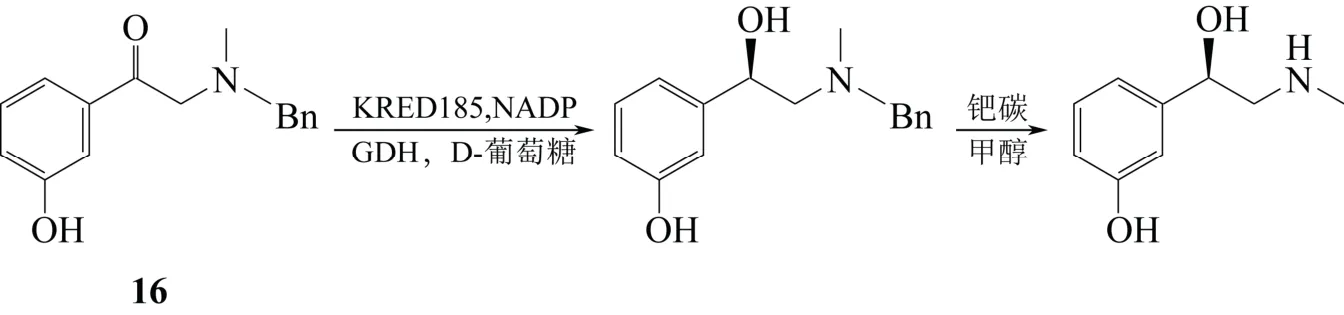
图16 酮还原酶催化合成路线

图17 醇脱氢酶催化合成路线

表2 生物不对称合成路线对比
3 结语与展望
随着医药行业的不断发展,L-去氧肾上腺素的各种新功能不断被开发,应用范围越来越广,需求量也不断增加。纵观L-去氧肾上腺素合成的历史,截止到2015年,经历了由拆分法到化学不对称合成法,再到生物不对称合成法的发展过程,并取得了很大的进展。
拆分法是合成L-去氧肾上腺素的传统工业方法,包括外消旋体的合成和外消旋体的拆分两部分,整体步骤繁杂,导致收率低、成本高,所得产品的光学纯度也比较低。Teerawutgulrag 提出的双键环氧化胺化以及双键的羟卤化加成胺化合成外消旋去氧肾上腺素的新路线(图4)简化了外消旋合成工艺。动态动力学拆分方法可用于L-去氧肾上腺素拆分工艺的改造(图9)。消旋体合成工艺的革新、动态动力学拆分等新技术的应用,将使拆分法在L-去氧肾上腺素的生产中继续发挥重要作用。
化学不对称合成法具有高催化活性、高原子经济性、高选择性的特点,但也需要结构复杂、含贵金属元素的手性催化剂,并且催化条件苛刻。因此探索更加温和的催化体系,采用价格便宜、环境友好的金属代替铑、钌等贵金属或开发不含金属元素的手性催化剂,并且实现催化剂的高效回收复用是未来发展的重要方向。若取得突破,该方法可成为最具经济性的方法,在未来将保持良好的发展态势。
生物不对称合成法步骤简单,反应条件温和,合成的产品具有很高的光学纯度,极具竞争性,是未来的一个发展方向。然而,该法尚处于研究的初期阶段,还存在着很多亟待解决的难题,如容量低、产率低、反应速度慢、后处理时存在相分离、酶的稳定性和使用寿命受限以及酶的回收再生等问题。随着这些难题的逐步解决,生物不对称合成法将逐步从实验室和基础研究阶段步入应用。
[1] Qin Y H,Guillory J K,Schoenwald R D. Formulation,in vitro dissolution,and ocular bioavailability of high- and low-melting phenylephrine oxazolidines[J]. Pharmaceutical Research,1993,10(11):1627-1631.
[2] Dewani A P,Dabhade S M,Bakal R L. Development and validation of a novel RP-HPLC method for simultaneous determination of paracetamol,phenylephrine hydrochloride,caffeine,cetirizine and nimesulide in tablet formulation[J]. Arabian Journal of Chemistry,2013,4(5):501-505.
[3] 王琰,陈文静,周颖,等. 纤维素衍生物手性固定相拆分肾上腺素、盐酸去氧肾上腺素[J]. 药物分析杂志,2012,32(11):1985-1990.
[4] 金有豫. 药理学[M]. 第5 版. 北京:人民卫生出版社,2001:39.
[5] Ma J,Wu L N,Hou Z,et al. Visualizing the endocytosis of phenylephrine in living cells by quantum dot-based tracking[J]. Biomaterials,2014,35:7042-7049.
[6] 于彩岩,王海,李晓光,等. 盐酸去氧肾上腺素光学异构体测定方法研究[J]. 内蒙古石油化工,2010(4):9-10.
[7] Dousa M,Gibala P,Havlicek J,et al. Drug-excipient compatibility testing-Identification and characterization of degradation products of phenylephrine in several pharmaceutical formulations against the common cold[J]. Journal of Pharmaceutical and BiomedicalAnalysis,2011,55:949-956.
[8] 倪永年,桂怿. 微分脉冲伏安法测定药物中盐酸去氧肾上腺素的含量[J]. 南昌大学学报,2009,33(1):35-37.
[9] Legerlotz H. Monohydric amino alcohols and their derivatives:DE,566578[P]. 1927-03-22.
[10] Legerlotz H. Optically active monohydroxyphenylalkylamines:DE,543529[P]. 1929-05-28.
[11] Legerlotz H. Optically active amino alcohols:DE,585164[P]. 1932-06-12.
[12] Legerlotz H. Beta-alkyl-amino compounds of mono-hydroxy-phenyl- ethanols and process of producing same:US,1932347[P]. 1933-10- 24.
[13] 马志聪. 一种去氧肾上腺素的制备方法:中国,101921197[P]. 2010-12-22.
[14] Dousa M,Gibala P,Havlicek J,et al. Drug-excipient compatibility testing-Identification and characterization of degradation products of phenylephrine in several pharmaceutical formulations against the common cold[J]. Journal of Pharmaceutical and Biomedical Analysis,2011,55:949-956.
[15] Bergamann E D,Sulzbacher M. A new synthesis of 1-(m- and p-hydroxyphenyl)-2-methylaminoethanol (m- and p-sympathol)[J]. Journal of Organic Chemistry,1951,16:84-89.
[16] Sun X Y,Rai R,Deschamps J R. Boc-protected 1-(3-oxocycloalkyl) ureas via a one-step Curtius rearrangement:Mechanism and scope[J]. Tetrahedron Letters,2014,55:842-844.
[17] Russel P B,Childress S J. New route to phenylephrine[J]. Journal of Pharmaceutical Sciences,1961,50:713-714.
[18] Hussain A A,Truelove J E,Lawrence,et al. Ester of 3-hydroxy-α- [(methylamino)methyl]benzyl alcohol:US,3825583[P]. 1974-07-23.
[19] Baison W,Teerawutgulrag A,Puangsombat P. An alternative synthesis of (±)-phenylephrine hydrochloride[J]. Maejo International Journal of Science and Technology,2014,8(01):41-47.
[20] Shimojuh N,Imura Y,Moriyama K,et al. Wittig reaction with ion-supported Ph3P[J]. Tetrahedron,2011,67:951-957.
[21] Bao W N,Pan H F,Zhang Z H,et al. Isolation of the stable strain Labrys sp. BK-8 for L-(+)-tartaric acid production[J]. Journal of Bioscience and Bioengineering,2014,118(1):1-5.
[22] 欧阳敬平,马国振,贾娴. 动态动力学拆分中外消旋化方法的研究进展[J]. 沈阳药科大学学报,2014,31(8):654-657.
[23] 李渤,李晓光,王晓红. L-去氧肾上腺素的制备方法:中国,103772215[P]. 2014-02-19.
[24] 杨少容,胡先明. 手性醇类化合物的构型翻转[J]. 化学试剂,2001,23(1):15-20.
[25] Prasad Divi M K,Krishna L M,Nageswara Rao B,et al. Process for resolution of 1-(3-hydroxyphenyl)-2-methylamino ethanol:US,8455692[P]. 2013-06-04.
[26] Tong S Q,Guan Y X,Yan J Z. Enantiomeric separation of (R,S)-naproxen by recycling high speed counter-current chromatography with hydroxypropyl-β-cyclodextrin as chiral selector[J]. Journal of Chromatography A,2011,1218:5434-5440.
[27] Gurjar M K,Krishna L M,Sarma B V N B S,et al. A practical synthesis of (R)-(-)-phenylephrine hydrochloride[J]. Organic Process Research & Development,1998,2(6):422-424.
[28] Larrow J F. Industrial applications of the jacobsen hydrolytic kinetic resolution technology[J]. Comprehensive Chirality,2012,9:129-146.
[29] Khan N H,Sadhukhan A,Maity N C,et al. Enantioselective O-acetylcyanation/cyanoformylation of aldehydes using catalysts with built-in crown ether-like motif in chiral macrocyclic V(Ⅴ) Salen complexes[J]. Tetrahedron,2011,67:7073-7080.
[30] 程咏梅. 化学酶法催化仲醇的动态动力学拆分[D]. 杭州:浙江大学,2010.
[31] 张怡. 氰醇及不饱和仲醇的动力学拆分研究[D]. 上海:东华大学,2011.
[32] Baumgarten W , Schiffers R. Process for manufacturing of enantiomerically pure 3-hydroxy-3-phenyl-propylamin : US ,7294744[P]. 2007-11-13.
[33] Takeda H,Tachinami T,Aburatani M,et al. Practical asymmetric synthesis of (R)-(-)-phenylephrine hydrochloride catalyzed by (2R,4R)-MCCPM-Rhodium complex[J]. Tetrahedron Letters,1989,30(3):367-370.
[34] Sakuraba S,Takahashi H,Takeda T,et al. Efficient asymmetric hydrogenation of α-amino ketone derivatives. A highly enantioselective synthesis of phenylephrine,levamisole,carnitine and propranolol[J]. Chemical and Pharmaceutical Bulletin,1995,43(5):738-747.
[35] Klingler F D,Wolter L,Dietrich W,et al.Verfahren zur herstellung von L-phenylephrin hydrochlorid:DE,19902229[P]. 2002-02-20.
[36] Hossein Michaelson. Synthesis of enantiomerically pure 2-hydroxy- 2-aryl-ethylamines:WO,2009086283[P]. 2008-11-22.
[37] McGarrity J F,Zanotti-Gerosa A. A feasibility study on the synthesis of phenylephrine via ruthenium-catalyzed homogeneous asymmetric hydrogenation[J]. Tetrahedron:Asymmetry,2010(21):2479-2486.
[38] 冯文化,郭方超,于晓丽. L-苯福林盐酸盐的制备方法:中国,102234237[P]. 2011-10-27.
[39] Xing X Y,Zhao Y H,Xu C,et al. Electronic helix theory-guided rational design of kinetic resolutions by means of the Sharpless asymmetric dihydroxylation reactions[J]. Tetrahedron,2012,68:7288-7294.
[40] Devi R,Borah R,Deka C R. Design of zeolite catalysts for Henry reaction under mild condition[J]. Applied Catalysis A:General,2012,433:122-127.
[41] Rajesh K P,Puspesh K U,Pradeep K. Enantioselective synthesis of (R)-phenylephrine hydrochloride[J]. Tetrahedron Letters,2003,44:6245-6246.
[42] Kureshy R I,Dangi B,Das A,et al. Recyclable Cu(Ⅱ)-macrocyclic salen complexes catalyzed Henry reaction of aldehydes:A practical strategy in the preparation of (R)-phenylephrine[J]. Applied Catalysis A:General,2012(439):74-79.
[43] Kureshy R I,Das A,Khan N H,et al. Cu(Ⅱ)-Macrocylic[H4]salen catalyzed asymmetric Henry reaction and its application in the synthesis of α1-adrenergic receptor agonist (R)-phenylephrine[J]. ACS Catalysis,2011,1(11):1529-1535.
[44] Alvizo O,Collier S J,Hennemann J,et al. Ketoreductase polypeptides for the preparation of phenylephrine: US ,20120149073[P]. 2012-06-14.
[45] Lin W D,Chen C Y,Chen H C,et al. Enantioselective synthesis of (S)-phenylephrine by whole cells of recombinant escherichia coli expressing the amino alcohol dehydrogenase gene from rhodococcus erythropolis BCRC 10909[J]. Process Biochemistry,2010,45:1529-1536.
[46] Peng G J,Cho Y C,Fu T K,et al. Enantioselective synthesis of (S)-phenylephrine by recombinant escherichia coli cells expressingthe short-chain dehydrogenase/reductase gene from serratia quinivorans BCRC 14811[J]. Process Biochemistry,2013,48:1509-1515.
[47] Peng G J,Kuan Y C,Chou H Y,et al. Stereoselective synthesis of (R)-phenylephrine using recombinant escherichia coli cells expressing a novel short-chain dehydrogenase/reductase gene from serratia marcescens BCRC 10948[J]. Journal of Biotechnology,2014,170:6-9.
[48] LenoX H J,Terentieva E,Gololobov M Y,et al. Optically active fiuorinated vasoconstrictors,methods for making them,and anesthetic formulations comprising them:US,6900203[P]. 2005-05-31.
[49] Tokoshima D,Hanaya K,Shoji M,et al. Whole-cell yeast-mediated preparation of (R)-2-chloro-1-(3-nitrophenyl)ethanol as a synthetic precursor for (R)-phenylephrine[J]. Journal of Molecular Catalysis B:Enzymatic,2013,97:95-99.
[50] Jin Z,Han S Y,Zhang L,et al. Combined utilization of lipase-displaying pichia pastoris whole-cell biocatalysts to improve biodiesel production in co-solvent media[J]. Bioresource Technology,2013,130:102-109.
[51] Navarro R,Ruiz P,Saucedo I,et al. Bismuth(Ⅲ) recovery from hydrochloric acid solutions using Amberlite XAD-7 impregnated with a tetraalkylphosphonium ionic liquid[J]. Separation and Purification Technology,2014,135:268-277.
[52] Jahani F,Tajbakhsh M,Golchoubian H,et al. Guanidine hydrochloride as an organocatalyst for N-Boc protection of amino groups[J]. Tetrahedron Letters,2011,52:1260-1264.
[53] Valizadeh H,Shomali A,Nourshargh S,et al. Carboxyl and nitrite functionalized graphene quantum dots as a highly active reagent and catalyst for rapid diazotization reaction and synthesis of azo-dyes under solvent-free conditions[J]. Dyes and Pigments,2015,113:522-528.
[54] 王波,孙勇,文军. 一种去氧肾上腺素的制备方法:中国,102776251[P]. 2012-08-21.
[55] Breuer M,Pletsch A,Hauer B,et al. Method for producing L-phenylephring using an alcohol dehydrogenase of aromaticum EBN1:US,8617854[P]. 2013-12-31.
