利用RNA-Seq技术鉴定拟南芥不定芽再生相关的转录因子
2015-07-19王兴春陈钊樊娟何苗苗韩渊怀杨致荣
王兴春,陈钊,樊娟,何苗苗,韩渊怀,杨致荣
利用RNA-Seq技术鉴定拟南芥不定芽再生相关的转录因子
王兴春1,2,陈钊3,樊娟1,何苗苗1,韩渊怀2,4,杨致荣3
1 山西农业大学生命科学学院,山西太谷 030801 2 山西农业大学农业生物工程研究所,山西太谷 030801 3 山西农业大学文理学院,山西太谷 030801 4 农业部黄土高原作物基因资源与种质创制重点实验室,山西太原 030031

王兴春, 陈钊, 樊娟, 等. 利用RNA-Seq技术鉴定拟南芥不定芽再生相关的转录因子. 生物工程学报, 2015, 31(4): 552–565.Wang XC, Chen Z, Fan J, et al. Identifying transcription factors involved in Arabidopsis adventious shoot regeneration by RNA-Seq technology. Chin J Biotech, 2015, 31(4): 552–565.
转录调控是不定芽再生过程的主要调控方式之一,但具体机制尚需进一步研究。为此,利用基于Illumina HiSeq™ 2000测序平台的RNA-Seq技术分析了不定芽再生缺陷突变体和野生型WS在愈伤形成和不定芽再生过程以及野生型WS从脱分化向再分化转变过程差异表达的转录因子 (Transcription factor,TF) 编码基因。结果表明:与野生型WS相比,在脱分化过程差异表达的TF编码基因有155个,其中表达量上调的97个,表达量下调的58个;在再分化过程差异表达的TF编码基因有68个,其中表达量上调的40个,表达量下调的28个;而在野生型WS从脱分化向再分化转变的过程,总共检测到231个差异表达的TF编码基因,包括160个表达量上调的基因和71个表达量下调的基因。其中,MYB-related (v-myb禽成髓细胞瘤病毒癌基因) 家族的不定芽相关转录因子基因在突变体脱分化阶段的表达量提高了3 217倍,是表达量上调最大的TF编码基因。进一步研究发现,该基因过量表达导致愈伤形成和不定芽再生缺陷,并抑制了幼苗特别是主根的生长发育,表明该基因是愈伤形成和不定芽再生过程的一个负调控因子。本研究不仅加深了人们对不定芽再生转录调控机制的认识,而且为今后不定芽再生相关转录因子的研究提供了大量候选基因信息。
不定芽再生,愈伤组织形成,RNA-Seq,转录因子,转录调控,拟南芥
作为植物再生完整植株的主要方式之一,不定芽再生不仅广泛应用于植物快速繁殖,而且是利用生物技术进行作物遗传改良的前提和基础[1]。植物激素尤其是细胞分裂素和生长素是影响不定芽再生的最主要因素。经典的拟南芥两步不定芽再生法即是通过调控这两种激素的比例来实现的:第一步,将拟南芥外植体在含有高浓度2,4-D的愈伤诱导培养基 (Callus induction medium,CIM) 上进行预培养。经过预培养,即可在外植体中柱鞘部位形成愈伤组织[2];第二步,将愈伤组织再转移到含有高浓度细胞分裂素的不定芽诱导培养基(Shoot induction medium,SIM) 继续培养,即可分化出不定芽[3]。因此,人们曾经乐观地认为只要合理调控细胞分裂素和生长素的比例,所有植物的组织或器官都能经过不定芽再生出完整植株。但时至今日,有些植物的离体再生仍非常困难,这极大地限制了基因功能的研究和重要农艺性状基因的应用。长期以来,人们普遍认为愈伤组织的形成是体细胞重编程从而回到一种未分化状态的过程。但Sugimoto等[4]的研究表明,各种器官形成的愈伤组织基因表达模式与根顶端分生组织的基因表达模式类似;而且愈伤组织和侧根的起始受到相同基因的调控,影响拟南芥侧根发育的基因同样也会影响植物离体器官发生。该研究从根本上改变了人们认为愈伤组织是未分化的细胞这一传统观念,使人们对不定芽再生机制有了更清晰的认识。
上述细胞分裂素和生长素调控不定芽再生的过程实质上是相关基因被激活或抑制的过程,在这一过程中转录因子起着极其重要的作用[5]。LBD (Lateral organ boundaries domain) 家族的、、和位于生长素信号下游,在CIM培养基上被迅速诱导表达[6]。这4个基因中的任何一个过量表达都会促进愈伤组织的形成;相反,功能缺失后导致愈伤形成能力受阻,表明LBDs转录因子是愈伤形成所必需的[6]。类似的,细胞分裂素早期响应的关键因子B型ARR在不定芽再生过程也起着关键的作用,而B型ARR是一类MYB转录因子[7-8]。APETALA2 (AP2) 家族的ESR1 (Enhancer of shoot regeneration 1) 是细胞分裂素信号下游的调控因子,过量表达及其同源基因可以促进不定芽的再生[9-11]。对和功能缺失突变体和的分析表明,单突变体不定芽再生能力都明显下降,而且双突变体不定芽再生能力比任何一个单突变体都差,暗示了这两个基因存在功能冗余[11]。AP2家族的另一成员的表达量在不定芽分化过程中显著增加,而该基因的突变则影响了芽分生组织特异基因的表达从而导致不定芽分化频率降低[12]。LFY (Leafy) 转录因子是一个调控植物从营养生长向生殖生长转变的关键因子[13]。当基因过量表达时,拟南芥根外植体可以直接再生出花器官,表明花发育相关基因在花器官离体再生过程也同样起着重要的作用[14]。尽管如此,人们对不定芽再生过程仍缺乏系统深入的认识。高通量测序技术的出现,使得大规模系统分离和鉴定不定芽再生相关转录因子成为可能[15-17]。
最近,我们在拟南芥中发现了一个编码糖苷水解酶13家族的() 基因,该基因的EMS点突变体愈伤形成和不定芽再生严重受阻[18]。为了阐明不定芽再生过程的转录调控机制,我们利用RNA-Seq技术检测了突变体与野生型在愈伤形成和不定芽再生两个阶段以及野生型从脱分化向再分化转变过程差异表达的基因,从中发现一大批相关的转录因子,并对其中一个负调控因子() 基因进行了深入研究。这些基因可作为不定芽再生转录调控机制研究的候选基因,其分离和鉴定将有助于全面解析不定芽再生调控网络。
1 材料与方法
1.1 植物材料和培养条件
RNA-Seq所用的拟南芥材料为Wassilewskija (WS) 野生型和突变体[18-19]。雌激素诱导的条件性过量表达转基因种子来自欧洲拟南芥种质中心 (European Arabidopsis Stock Centre,NASC) 的TRANSPLANTA种质[20]。除非特别说明,拟南芥种子均播种于1/2 MS培养基:2.3 g/L MS基本盐 (Murashige and Skoog Basal Medium w/Vitamins,PhytoTechnology Laboratories,货号M519)、2%蔗糖和0.8%的琼脂粉,pH值5.8。拟南芥幼苗和外植体生长温度均为22 ℃。采用T8 LED灯管照明,灯管红、蓝、橙和白光灯珠的比例6∶2∶1∶1。光周期为16 h光照/8 h黑暗交替,光照强度为80−120 µmol/(m2·s)。
1.2 愈伤组织和不定芽的诱导
愈伤组织和不定芽的诱导参照王兴春等[17-18]的方法进行:用剪刀剪取20−30 µmol/(m2·s)弱光下培养的拟南芥下胚轴,置于CIM培养基诱导愈伤,7 d后再转移到SIM培养基诱导不定芽。CIM和SIM的配方详见王兴春等[17-18]的报道。
1.3 总RNA的提取和RNA-Seq高通量测序及测序数据分析
RNA-Seq实验材料为在CIM培养7 d的WS (命名为WS-CIM7) 和(命名为-CIM7) 以及在SIM培养2 d的WS (命名为WS-SIM2) 和(命名为-SIM2)。为了提高实验结果的准确性并节约测序费用,我们进行了3次生物学重复。3次重复的样品分别提取RNA,然后等量混合用于RNA-Seq测序,测序反应仅进行1次。首先称取100 mg外植体材料,将其放入装有液氮的研钵中,快速研磨成粉末;然后利用康为世纪公司含DNaseⅠ的植物RNA提取试剂盒 (货号CW0559) 提取总RNA,提取步骤按照试剂盒说明书进行。RNA-Seq高通量测序利用Illumina HiSeq™ 2000测序仪进行。测序产生的图像数据首先经碱基识别 (Base calling) 转为原始碱基序列数据,然后去除含有接头的序列和低质量的序列,产生的数据称之为有效读段 (Clean reads)。对有效读段进行测序质量评估、比对统计、测序饱和度分析和在参考基因上的分布分析,全部分析合格的即可用于基因的差异表达分析。基因表达量的计算采用RPKM法进行[21],根据两个样本间基因的RPKM值来筛选差异表达的基因。差异表达基因的筛选参照Audic等[22]的方法进行,将错误发现率 (False discovery rate,FDR) ≤0.001且差异倍数不低于2倍即|log2|≥1的视为差异表达基因。
1.4 不定芽再生相关转录因子的筛选和分类
为了筛选差异表达的转录因子,我们将所有差异表达的基因映射到Gene ontology (http://www.geneontology.org/) 数据库进行基因注释 (Gene annotation),然后根据其参与的生物过程 (Biological process) 进行分类,GO term为0001071具有核酸结合转录因子活性的即为不定芽再生相关的转录因子。所筛选到的转录因子根据PlantTFDB 3.0[23]进行分类。
1.5 雌激素诱导基因过量表达
愈伤形成阶段雌激素诱导及观察方法:在CIM培养基中添加10 μmol/L的17 β-雌二醇 (货号E-8875,Sigma-Aldrich),培养7 d。然后,利用Olympus BX51显微镜观察拍照。不定芽再生阶段雌激素诱导方法:弱光培养的拟南芥下胚轴在无17 β-雌二醇的CIM培养基上培养7 d,然后再转移到含有10 μmol/L的17 β-雌二醇的SIM培养基继续培养。拟南芥幼苗的雌激素诱导方法:将拟南芥种子播种在含有不同浓度 17 β-雌二醇的1/2 MS培养基,4 ℃低温春化2 d,取出后置于拟南芥培养间培养。
2 结果与分析
2.1 RNA-Seq测序结果评估
为了解析突变体在愈伤形成和不定芽再生两个阶段以及野生型WS从脱分化向再分化转变过程差异表达的TF编码基因,我们选取了在CIM培养7 d的WS (WS-CIM7) 和(-CIM7) 以及在SIM培养2 d的WS (WS-SIM2) 和(-SIM2) 进行RNA-Seq分析。经测序WS-CIM7、-CIM7、WS-SIM2和-SIM2 4个样品分别得到10 184 656、10 898 770、9 399 484和9 788 169条有效读段 (表1和表2,以及王兴春等[17]的数据,NCBI Sequence Read Archive accession No.分别为SRR1144842、SRR1144843、SRR1144844、和SRR1144845),其有效读段分别占99.59%、99.55%、99.55%和99.56%。然后,将WS-CIM7、-CIM7、WS-SIM2和-SIM2样品的有效读段分别与拟南芥基因数据库进行比对分析,结果表明匹配读段 (Total mapped reads) 分别占92.96%、92.90%、93.13%和93.02% (表1和表2,以及王兴春等[17]的数据)。测序饱和度分析表明,这4个样品检测到的基因数在测序量较小时均随着测序量的增加而增加;当测序量达到4 M时,其增长趋于平缓;而当测序量达到8 M时,检测到的基因数趋于饱和 (图1A和1B,以及王兴春等[17])。而这4个样品的测序量均在9 M以上,因此可以认为测序基本覆盖细胞中表达的全部基因,具有代表性。随后,我们又分析了有效读段在拟南芥参考基因上的分布情况,由图1C和1D 以及王兴春等[17]的数据可知这4个样品的有效读段分布均匀性较好,表明mRNA是随机打断的。综上所述,这4个样品测序质量较高,可用于下一步差异表达基因的研究。
2.2 愈伤形成过程差异表达的TF编码基因
对CIM培养基上培养7 d的WS和外植体的RNA-Seq数据进行差异比较分析,共筛选出1 860个差异表达的基因,其中TF编码基因为155个,约占总差异表达基因的8.3% (表3)。这155个TF编码基因中,外植体中表达量上调的有97个,表达量下调的有58个。进一步研究发现,这155个转录因子属于26个家族,主要涉及激素信号转导、胁迫响应和根毛或侧根的发生以及生长发育的调控等过程[24-26]。值得一提的是,基因的突变导致(AT1G24590) 基因的表达量下降了5.05倍。该基因在不定芽再生过程起着重要的作用[11],但对脱分化的影响还需要进一步研究。
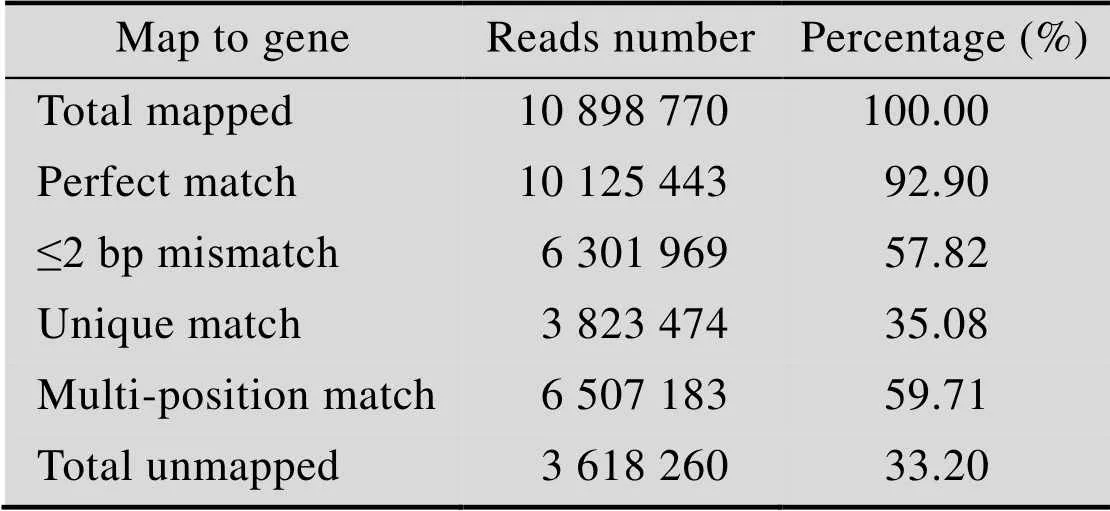
表1 样品be1-CIM7测序读段与拟南芥参考基因的比对统计结果

表2 样品be1-SIM2测序读段与拟南芥参考基因的比对统计结果

图1 RNA-Seq测序质量评估
2.3 不定芽再生早期差异表达的TF编码基因
为了解析不定芽再生早期的转录调控机制,我们比较了野生型WS拟南芥外植体从脱分化 (样品WS-CIM7) 向再分化 (样品WS-SIM2) 转变过程差异表达的基因。如表4所示,共检测到231个差异表达的TF编码基因,其中表达量下调的为71个,表达量上调的有160个。值得注意的是,和基因的表达量分别上调了7.59倍和2.25倍,而这两个基因已经被证明是不定芽再生过程的关键基因[9,12]。除此之外,NAC家族的() 和的表达量也大幅上调,分别上调了8.94倍和5.09倍,这一结果与Che等[12]的研究是一致的。
为了深入了解不定芽再生早期的转录调控机制并解析突变体不定芽再生障碍的原因,我们进一步比较了在SIM培养基上培养2 d的(SIM2) 和WS (WS-SIM2) 外植体中差异表达的基因。共检测到832个差异表达基因,其中68个基因编码转录因子,占8.17% (表5)。这些转录因子涉及20个家族,其中数目最多的为NAC家族、ERF家族和WRKY家族,分别为13个、9个和8个 (表5)。
2.4 过量表达基因抑制了不定芽再生
由于拟南芥突变体中柱鞘发育异常,从而导致外植体愈伤组织形成受阻[18]。我们推测,外植体愈伤形成缺陷的表型可能与基因的转录调控有关。为了证实这一推测,并验证所筛选转录因子的真实性,我们从表3中选取了一个MYB-related家族的AT3G10590基因进行了深入研究。该基因在突变体和野生型外植体中的转录本数分别为13和0,RPKM值分别为3.217和0.001,表达量上升了3 217倍,是所有基因中上调倍数最大的一个,故将其命名为() 基因。此外,我们从NASC获得了基因的两个雌激素诱导过量表达的转基因株系TPT_3.10590.1F和TPT_3.10590.1I。由于TPT_3.10590.1F和TPT_3.10590.1I在所有的实验中表型类似,只是TPT_3.10590.1I的表型比TPT_3.10590.1F的更强,因此在本文仅给出TPT_3.10590.1I的结果,并将其命名为(ART1 overexpression)。

表3 愈伤形成过程be1-3与WS野生型相比差异表达的TF编码基因
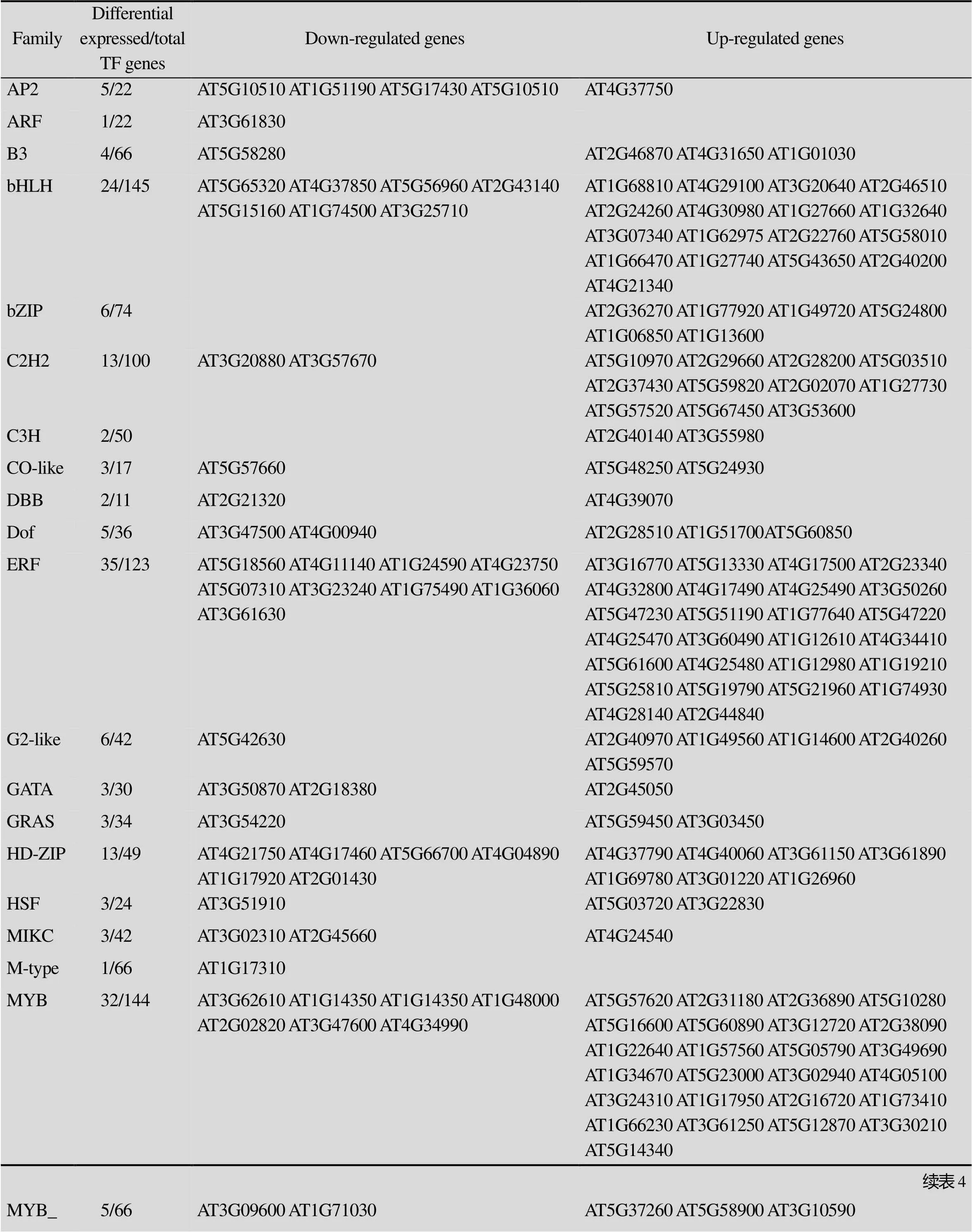
表4 野生型拟南芥外植体从脱分化向再分化转变过程差异表达的TF编码基因
为阐明在不定芽再生过程的作用,分别将和Col-0野生型的下胚轴培养在0和10 μmol/L雌二醇的CIM培养基上。培养7 d后,无论是在0还是10 μmol/L雌二醇CIM培养基上培养的Col-0野生型下胚轴顶端和中柱都已经膨大形成愈伤组织,且二者无明显差异 (图2A和2B),表明雌二醇对愈伤的形成无显著影响。与野生型类似,无雌二醇CIM上培养的外植体顶端因形成大量愈伤而膨大成球状 (图2C),而雌二醇培养基上培养的外植体顶端仅稍微变粗 (图2D)。类似的,外植体中柱鞘部位在无雌二醇时膨大形成了突起 (图2E),而在雌二醇培养基上的则无明显变化 (图2F)。然后,将无雌二醇CIM培养的外植体分别转到0和10 μmol/L雌二醇的SIM培养基继续培养。14 d后,无论是否有雌二醇,野生型外植体都可以分化出绿芽 (图2G和2H),但仅在无雌二醇的培养基上能分化出绿芽,在雌二醇培养基上培养时仅有少数绿点 (图2I和2J)。这表明,基因是愈伤形成和不定芽再生过程的一个负调控因子。
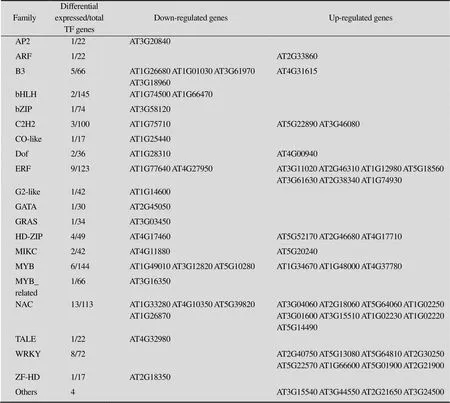
表5 不定芽再生早期be1-3中差异表达的TF编码基因
2.5 过量表达基因抑制了幼苗的生长发育
上述结果是在离体条件下获得的,为解析基因在幼苗生长和发育中作用,将播种在不同浓度的17 β-雌二醇培养基上。结果表明,在无诱导剂的培养基上,幼苗生长发育完全正常 (图3A),然而在雌激素培养基上,幼苗生长发育缓慢,根系变短 (图3B–3F)。而且诱导剂的浓度越高,这种表型也越严重 (图3B–3F)。在10 μmol/L 17 β-雌二醇培养基上,的主根不能伸长,而且长出大量毛状根系 (图3E和3F)。这表明,基因在植物生长发育和离体器官再生过程都起着重要的作用。

图2 过量表达ART1基因抑制了不定芽再生
3 讨论
本研究利用RNA-Seq高通量测序技术检测了脱分化和再分化过程以及从脱分化向再分化转变过程差异表达的TF编码基因,获得了不定芽再生过程转录调控的重要信息。拟南芥基因组中共有1 716个TF编码基因,分属于58个转录因子家族[23]。在拟南芥不定芽再生过程,共检测到341个差异表达的TF编码基因 (表3−5),占拟南芥总TF编码基因的19.87%。这341个转录因子分属于29个家族,其中ERF家族是差异表达的TF编码基因最多的家族,其次是MYB家族和NAC家族,这3个家族差异表达的基因数分别为50、40和38个,分别占差异表达转录因子的14.67%、11.73%和11.14% (表3−5)。ERF家族是拟南芥中的一个转录因子大家族,有123个成员,占拟南芥转录因子总数的7.17%;而在本研究中差异表达的占总差异表达TF编码基因的14.67%,富集了2.05倍。的富集可能与该家族广泛参与了激素信号转导、胁迫响应和生长发育调控等有关[27]。这暗示了与其他家族的转录因子相比,ERF家族的转录因子在不定芽再生过程可能起着更为重要的作用。进一步分析发现,外植体在愈伤形成和不定芽再生两个阶段表达模式一致 (均上调或下调) 的TF编码基因有26个,表达模式不一致的TF编码基因有5个 (表3和表5)。这26个表达模式一致的基因可能在这两个过程都起作用,是不定芽再生转录调控的关键TF编码基因。
在本研究中,我们发现了一些已知的与不定芽再生有关的TF编码基因,这些基因包括、和等[9,11-12]。其中,在野生型拟南芥外植体从脱分化向再分化转变时基因的表达量上调了7.59倍;而在外植体脱分化阶段基因的表达量下调了5.05倍,这与其功能是相适应的。这表明本实验反映了拟南芥不定芽再生过程基因差异表达的真实情况,结果是可信的。在脱分化过程,与野生型相比突变体中一系列与根系发育相关的基因差异表达 (表3),而根系尤其是侧根的发育与愈伤的形成有着密切的关系[4],深入研究这些基因的功能有望全面揭示愈伤形成和根系发育的关系。Fan等[6]的研究表明,、和等4个基因在愈伤形成过程起着重要的调控作用,但在本实验中未检测到这4个基因的差异表达。这可能是因为基因是一个碳水化合物代谢相关的基因,位于愈伤形成调控网络的下游,因此其突变不会影响到上游基因的表达。不过,相对于已有的数据,本研究挖掘了更为丰富的拟南芥不定芽再生相关的转录因子信息,为今后深入研究不定芽再生转录调控机制提供了重要的参考。
拟南芥基因通过碳水化合物代谢调控了植物生长发育和离体器官再生[18-19],但在愈伤形成和不定芽再生两个阶段均未发现直接参与碳水化合物代谢的转录因子。其原因可能有3个:1)基因编码一个糖苷水解酶,位于碳水化合物代谢的下游,其变化对这两个过程中其他碳水化合物代谢影响很小;2) CIM和SIM培养基均添加了大量外源碳水化合物,这也可能在一定程度上影响了由于缺失导致的相关基因表达的程度;3) 已发现的转录因子中,大部分功能未知。在愈伤形成过程,外植体中表达量上调的TF编码基因有97个,下调的TF编码基因有58个 (表3)。外植体愈伤形成缺陷的表型可能就是由于这些基因表达变化导致的。进一步实验表明,表达量上调倍数最大的基因是愈伤形成的一个负调控因子,其过量表达导致愈伤形成和不定芽再生严重受阻。但这并不意味着基因过量表达是导致突变体愈伤形成缺陷的唯一原因。要证明这一推测,可以构建的双突变体,进一步分析愈伤形成情况。若双突变体再生能力与野生型相同,则表明基因过量表达是导致突变体愈伤形成缺陷的主要原因,否则表明不定芽再生缺陷是多种原因造成的。此外,基因突变导致基因表达量大幅上调,表明基因负调控基因的表达。但这种调控不是直接的,可能是由于基因的代谢产物 (例如碳水化合物) 抑制了基因的表达。
REFERENCES
[1] Guan CM, Zhang XS. Advances in the molecular mechanism ofplant organogenesis. Chin Bull Bot, 2006, 23(5): 595−602 (in Chinese).关春梅, 张宪省. 植物离体器官发生控制机理研究进展. 植物学通报, 2006, 23(5): 595−602.
[2] Atta R, Laurens L, Boucheron-Dubuisson E, et al. Pluripotency ofxylem pericycle underlies shoot regeneration from root and hypocotyl explants grown. Plant J, 2009, 57(4): 626−644.
[3] Valvekens D, Montagu MV, Van Lijsebettens M. Agrobacterium tumefaciens-mediated transformation ofroot explants by using kanamycin selection. Proc Natl Acad Sci USA, 1988, 85(15): 5536−5540.
[4] Sugimoto K, Jiao Y, Meyerowitz EM.regeneration from multiple tissues occurs via a root development pathway. Dev Cell, 2010, 18(3): 463−471.
[5] Long TA, Benfey PN. Transcription factors and hormones: new insights into plant cell differentiation. Curr Opin Cell Biol, 2006, 18(6): 710−714.
[6] Fan M, Xu C, Xu K, et al. LATERAL ORGAN BOUNDARIES DOMAIN transcription factors direct callus formation inregeneration. Cell Res, 2012, 22(7): 1169−1180.
[7] Sakai H, Honma T, Aoyama T, et al. ARR1, a transcription factor for genes immediately responsive to cytokinins. Science, 2001, 294(5546): 1519−1521.
[8] Hill K, Mathews DE, Kim HJ, et al. Functional characterization of type-B response regulators in thecytokinin response. Plant Physiol, 2013, 162(1): 212−224.
[9] Banno H, Ikeda Y, Niu QW, et al. Overexpression ofinduces initiation of shoot regeneration. Plant Cell, 2001, 13(12): 2609−2618.
[10] Ikeda Y, Banno H, Niu QW, et al. Thegene inregulatesat the transcriptional level and controls cotyledon development. Plant Cell Physiol, 2006, 47(11): 1443−1456.
[11] Matsuo N, Makino M, Banno H.()andregulateshoot regeneration and their expressions are differentially regulated. Plant Sci, 2011, 181(1): 39−46.
[12] Che P, Lall S, Nettleton D, et al. Gene expression programs during shoot, root, and callus development intissue culture. Plant Physiol, 2006, 141(2): 620−637.
[13] Weigel D, Alvarez J, Smyth DR, et al.controls floral meristem identity in. Cell, 1992, 69(5): 843−859.
[14] Wagner D, Wellmer F, Dilks K, et al. Floral induction in tissue culture: a system for the analysis of LEAFY-dependent gene regulation. Plant J, 2004, 39(2): 273−282.
[15] Gliwicka M, Nowak K, Balazadeh S, et al. Extensive modulation of the transcription factor transcriptome during somatic embryogenesis in. PLoS ONE, 2013, 8(7): e69261.
[16] Wang XC, Yang ZR, Wang M, et al. High-throughput sequencing technology and its application. China Biotechnol, 2012, 32(1): 109−114 (in Chinese).王兴春, 杨致荣, 王敏, 等. 高通量测序技术及其应用. 中国生物工程杂志, 2012, 32(1): 109−114.
[17] Wang XC, Yang ZR, Zhang SW, et al. Digital gene expression profiling analysis of the early adventitious shoot formation inChin J Biotech, 2013, 29(2): 189−202 (in Chinese).王兴春, 杨致荣, 张树伟, 等. 拟南芥不定芽发生早期的数字基因表达谱分析. 生物工程学报, 2013, 29(2): 189−202.
[18] Wang XC, Yang ZR, Wang M, et al. Thegene, encoding a glycoside hydrolase family 13 protein, is required forplant regeneration in. Plant Cell Tiss Org Cult, 2014, 117(2): 279−291.
[19] Wang XC, Xue L, Sun JQ, et al. Thegene, encoding a putative glycoside hydrolase localized in plastids, plays crucial roles during embryogenesis and carbohydrate metabolism. J Integr Plant Biol, 2010, 52(3): 273−288.
[20] Coego A, Brizuela E, Castillejo P, et al. The TRANSPLANTA collection oflines: a resource for functional analysis of transcription factors based on their conditional overexpression. Plant J, 2014, 77(6): 944−953.
[21] Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods, 2008, 5(7): 621−628.
[22] Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res, 1997, 7(10): 986−995.
[23] Jin J, Zhang H, Kong L, et al. PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res, 2014, 42(Database issue): D1182−1187.
[24] Yi K, Menand B, Bell E, et al. A basic helix-loop-helix transcription factor controls cell growth and size in root hairs. Nat Genet, 2010, 42(3): 264−267.
[25] Karas B, Amyot L, Johansen C, et al. Conservation of lotus andbasic helix-loop-helix proteins reveals new players in root hair development. Plant Physiol, 2009, 151(3): 1175−1185.
[26] Sakuma Y, Maruyama K, Qin F, et al. Dual function of antranscription factor DREB2A in water-stress-responsive and heat-stress-responsive gene expression. Proc Natl Acad Sci USA, 2006, 103(49): 18822−18827.
[27] Nakano T, Suzuki K, Fujimura T, et al. Genome-wide analysis of thegene family inand rice. Plant Physiol, 2006, 140(2): 411−432.
(本文责编陈宏宇)
Identifying transcription factors involved inadventious shoot regeneration by RNA-Seq technology
Xingchun Wang1,2, Zhao Chen3, Juan Fan1, Miaomiao He1, Yuanhuai Han2,4, and Zhirong Yang3
1,,030801,,2,,030801,,3,,030801,,4,,030031,,
Transcriptional regulation is one of the major regulations in plant adventious shoot regeneration, but the exact mechanism remains unclear. In our study, the RNA-seq technology based on the Illumina HiSeq™ 2000 sequencing platform was used to identify differentially expressed transcription factor (TF) encoding genes during callus formation stage and adventious shoot regeneration stage between wild type and adventiousshoot formationdefective mutantand during the transition from dedifferentiation to redifferentiation stage in wildtype WS. Results show that 155 TFs were differentially expressed betweenmutant and wild type during callus formation, of which 97 genes were up-regulated, and 58 genes were down-regulated; and that 68 genes were differentially expressed during redifferentiation stage, with 40 genes up-regulated and 28 genes down-regulated; whereas at the transition stage from dedifferentiation to redifferention in WS wild type explants, a total of 231 differentially expressed TF genes were identified, including 160 up-regualted genes and 71 down-regulated genes. Among these TF genes, the adventious shoot related transcription factor 1 () gene encoding a MYB-related (v-myb avian myeloblastosis viral oncogene homolog) TF, was up-regulated 3 217 folds, and was the highest up-regulated gene duringcallus formation. Over expression of thegene caused defects in callus formation and shoot regeneration and inhibited seedling growth, indicating that thegene is a negative regulator of callus formation and shoot regeneration. This work not only enriches our knowledge about the transcriptional regulation mechanism of adventious shoot regeneration, but also provides valuable information on candidate TF genes associated with adventious shoot regeneration for future research.
adventious shoot regeneration, callus formation, RNA-Seq, transcription factor, transcriptional regulation,
November 4, 2014; Accepted: December 11, 2014
Xingchun Wang. Tel: +86-354-6287191-307; E-mail: wxingchun@163.com Zhirong Yang. Tel: +86-354-6288341; E-mail: zryangsx@163.com
Supported by:National Natural Science Foundation of China (No. 31100235), Natural Science Foundation of Shanxi (No. 2013011028-1), Shanxi Scholarship Council of China (No. 2010050).
国家自然科学基金(No. 31100235),山西省自然科学基金 (No. 2013011028-1),山西省回国留学人员科研资助项目(No. 2010050) 资助。
网络出版时间:2015-02-03
http://www.cnki.net/kcms/detail/11.1998.q.20150203.1622.003.html
