慢性阻塞性肺疾病炎症机制研究进展
2015-07-19复综述罗治海审校
周 复综述,罗治海审校
(重庆市巴南区人民医院呼吸科401320)
慢性阻塞性肺疾病炎症机制研究进展
周 复综述,罗治海审校
(重庆市巴南区人民医院呼吸科401320)
肺疾病,慢性阻塞性; 炎症; 细胞因子; 综述
慢性阻塞性肺疾病(COPD)在世界范围内患病率及病死率都日渐增高,给经济和社会带来了巨大的负担,全球疾病负担研究项目(GBDS)和世界卫生组织(WHO)预测,到2020年,COPD将成为世界第三大死亡原因和第五大世界经济负担。COPD全球倡议(GOLD,updated 2014)对COPD的定义是:COPD是一种普遍可以预防和治疗的疾病,以持续性的气流受限为特征,通常呈进行性发展与呼吸道和肺对有害颗粒(或气体)的慢性炎性反应增强有关[1]。COPD发病机制仍不清楚,目前公认的机制包括炎性反应机制、氧化/抗氧化失衡、蛋白酶/抗蛋白酶失衡机制等,其中炎性反应机制被认为是最重要的。本文对COPD炎症致病机制中较重要的炎症细胞和炎症介质进展进行综述。
1 COPD与炎性反应
炎症机制认为,有害气体或颗粒中以烟草危害最为常见,进入呼吸道后引起机体局部炎症级联反应,分泌各种细胞因子和趋化因子,激活炎症细胞(包括巨噬细胞、T淋巴细胞、B淋巴细胞、中性粒细胞等)和结构细胞(包括上皮细胞、内皮细胞和成纤维细胞等),释放出各种炎症介质和酶类破坏正常呼吸道结构,反复炎症导致COPD临床症状出现,尤以肺气肿和慢性支气管炎为主。如图1所示,巨噬细胞在COPD发病机制起中心作用,当烟草或其他有害颗粒被吸入气道后刺激巨噬细胞释放各种炎症介质,启动COPD发生;中性粒细胞可能被趋化因子生长调节基因1(CXCL-1)、CXCL-8、白介素-8(IL-8)、白三烯B4(LTB4)等趋化物质吸引,进一步放大炎性反应;蛋白酶[基质金属蛋白酶-2(MMP-2)、MMP-9、MMP-12、中性粒细胞弹性蛋白酶、组织蛋白酶G、蛋白酶3、MMP-8、组织蛋白酶-K、组织蛋白酶-L和组织蛋白酶-S等]和CD8+淋巴细胞共同作用导致正常呼吸道结构破坏和黏液高分泌,出现肺气肿型和支气管炎型临床表现;转化生化因子-β(TGF-β)的释放可能诱导小气道纤维化。
2 炎症细胞与炎症介质
2.1 炎症细胞
2.1.1 上皮细胞 被激活的上皮细胞产生的炎症介质,包括肿瘤坏死因子-α(TNF-α)、IL-1β、IL-6、粒细胞-巨噬细胞集落刺激因子(GM-CSF)、IL-8、TGF-β、血管内皮生长因子(VEGF)及IL-33等。IL-33水平在COPD患者血清及外周血淋巴细胞均有升高,烟草提取物和脂多糖刺激都能提高淋巴细胞和支气管上皮细胞表达IL-33的能力,免疫荧光证实肺组织的IL-33主要来自支气管上皮细胞[2]。VEGF能够维持正常肺泡结构的完整性,小鼠模型研究结果显示,阻断VEGF受体能够诱导细胞的凋亡和肺气肿样改变[3]。
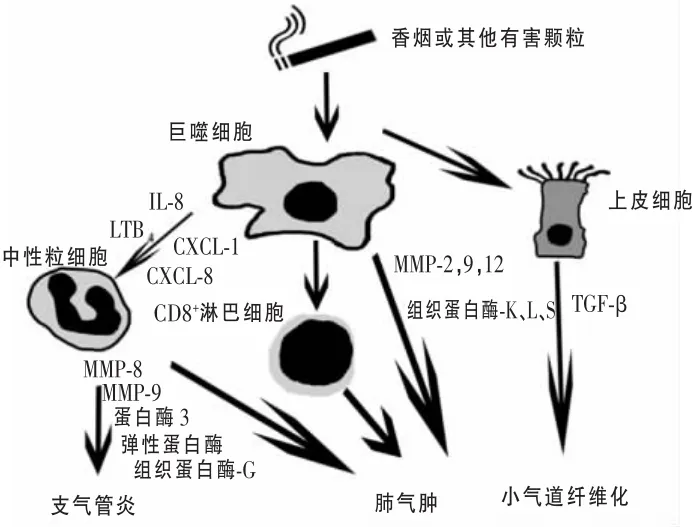
图1 COPD的炎症机制
2.1.2 巨噬细胞 在COPD发生进展中,巨噬细胞是最重要的细胞之一,尤其在慢性炎症的启动阶段具有决定性作用,也是研制COPD抗感染治疗药物的主要靶细胞。巨噬细胞不仅在COPD患者的痰、支气管肺泡灌洗液、气管和肺实质细胞中明显增多,而且在破坏的肺泡中也出现聚集[4]。吸入有害物质后,巨噬细胞在气管内和肺组织内发生聚集,激活后释放TNF-α、IL-1、IL-8、单核细胞趋化蛋白-1(MCP-1)、LTB4、活性氧(ROS);而肺泡中的巨噬细胞还会分泌一些弹性蛋白水解酶MMP-2、MMP-9、MMP-12和组织蛋白酶-K、组织蛋白酶-L、组织蛋白酶-S[5]。
2.1.3 中性粒细胞 研究发现,COPD患者的痰及支气管肺泡灌洗液的中性粒细胞数量明显增多,然而在呼吸道和肺实质中性粒细胞数量增加得却不是很明显,推测可能是中性粒细胞快速地通过了呼吸道和肺实质所致[6]。激活的上皮细胞和巨噬细胞在气道中分泌LTB4、IL-8、趋化因子CXCL-1和CXCL-8、上皮中性粒细胞活化肽-78(ENA-78)等,在这些趋化因子作用下募集大量的中性粒细胞,活化的中性粒细胞释放出中性粒细胞弹性蛋白酶、组织蛋白酶-G、蛋白酶3、MMP-8、MMP-9等,这些蛋白酶会破坏正常肺组织并强有力地促进支气管黏液的分泌[5],其中的IL-8也能够由活化的中性粒细胞分泌。推测中性粒细胞进一步放大了炎性反应,促进了COPD的形成和发展。
2.1.4 T淋巴细胞 淋巴细胞浸润在COPD发病机制中被认为是最关键的因素之一,无论是气道还是肺实质T淋巴细胞的总量都明显增加,以CD8+T淋巴细胞增加为主,增加量与肺泡破坏和呼吸道的阻塞程度密切相关。目前有学者认为,CD4+T淋巴细胞与疾病的严重性相关,CD8+T淋巴细胞与肺组织破坏相关[7]。有研究发现,COPD患者气道中CD8+T细胞通过分泌颗粒酶B、穿孔素等诱导细胞凋亡和DNA裂解,加重COPD病情进展[8]。有研究认为这可能是由于增加的T细胞中有部分出现免疫衰退,不能表达CD28受体,导致穿孔素和颗粒酶B分泌增加[9]。虽然吸烟的COPD患者气道中CD4+细胞增加的数量不多,但这部分细胞能够表达活化的转录激活因子4(STAT-4),该转录因子对于激活和调节1型辅助性T细胞(Th1)细胞谱系的发育是必不可少的[10]。
2.1.5 嗜酸性粒细胞 10%~40%的COPD患者可发现诱导痰中嗜酸性粒细胞增多,痰中嗜酸性粒细胞不仅可用于评估COPD患者对糖皮质激素的治疗敏感性,而且通过降低气道中嗜酸性粒细胞数量会明显降低COPD急性加重的发生率[11]。外周血中嗜酸性粒细胞与支气管黏膜下的嗜酸性粒细胞存在联系,二者与支气管网状基底膜增厚密切相关,被认为是气道重塑的原因之一[12]。
2.2 炎症介质 目前认为,在COPD发病中起重要作用的介质有脂类介质(LTB-4、前列腺素E2、前列腺素F2α)、趋化因子(CXCL-1、CXCL-8、CXCL-9、CXCL-10、CXCL-11、CCL-2、CCL5)、细胞因子(TNF-α、IL-1β、IL-6、IL-8、IL-17)、生长因子等,有些因子来源于激活的炎症细胞,有些来源于肺实质细胞,COPD的发展不是一种炎症介质单一作用的结果,而是这些因子相互作用所致,其作用范围也不局限于肺组织,还可能会造成身体其他部位损害或加重一些并发症[13]。
2.2.1 酯类炎性介质 (1)LTB4:COPD的重要特征是中性粒细胞浸润,中性粒细胞在浸润部位通过释放趋化物质招募更多的白细胞聚集,进一步放大炎性反应[14],LTB4被认为是炎症放大过程中早期释放的重要趋化因子[15],能够明显地提高中性粒细胞招募数量和促进中性粒细胞向炎症中心部位迁移。Afonso等[16]研究发现,LTB4的合成放大了甲酰肽介导的中性粒细胞极化和趋化,推测LTB4可能是细胞间远距离信号传递分子,能够调节中性粒细胞的迁移和招募。有研究结果显示,COPD患者诱导痰和呼出气冷凝物中均检出LTB4,尤其在COPD急性加重期,LTB4浓度增加更显著[17]。(2)前列腺素E2(PGE2):是一种重要的炎症介质,环氧合酶2是PGE2合成的限速酶,PGE2的合成可能会促进肺部成纤维细胞衰老及炎症进展[18]。此外,有研究发现,COPD患者诱导痰中PGE2增加程度与气流受限程度是一致的,推测PGE2可能通过MMP-2介导参与COPD炎症进展及气道重塑[19]。
2.2.2 细胞因子 (1)TNF-α:TNF-α是一种重要的中性粒细胞趋化物,由单核巨噬细胞系和肥大细胞分泌,COPD稳定期和急性加重期患者的诱导痰中均出现明显增加。有研究发现,给予健康人和老鼠雾化吸入重组人TNF-α制剂或使用抗原、细菌内毒素、呼吸道病毒等刺激物产生内源性TNF-α,均能诱导中性粒细胞和嗜酸性粒细胞浸润,导致细胞破坏、黏液化生、气道纤维化及气道高反应[20]。(2)IL-1:主要由单核巨噬细胞和成纤维细胞分泌,作用于白介素-1受体1(IL-1R1),有证据显示其参与肺气肿和气道炎症的发生[21-22],尤其在COPD急性加重期会明显升高。Sichelstiel等[23]在流感病毒诱导COPD急性加重期的模型中发现,流感病毒感染早期(24 h内),IL-1β需要IL-17A介导才会引起中性粒细胞趋化,而在感染后期,IL-1β可以单独诱导中性粒细胞浸润并进一步放大炎性反应。(3)IL-6:COPD患者血浆和诱导痰中同时出现IL-6水平升高,且升高程度与肺功能损伤程度有关,IL-6与干扰素-γ(INF-γ)、IL-1β和IL-1受体拮抗剂多态性共同作用可引起吸烟人群肺功能下降[24]。IL-6基因位于人类第7号染色体,存在多个基因多态性,在吸烟人群中,IL-6-174G/C单核苷酸基因多态性与第1秒用力呼气容积(FEV1)的快速下降和COPD发病风险相关[25]。IL-6作为一种重要的炎症标记物,与白细胞计数、C反应蛋白(CRP)、IL-8、纤维蛋白原、趋化因子CCL-18/PARC和表面活性蛋白-D(SP-D)共同用于预测COPD死亡率,明显地提高了预测的准确性。该试验是经大样本、多中心、长时间的随访得出的结果,对临床具有重要的指导性[26]。(4)IL-17:IL-17家族有A~E 6个同源的细胞因子,研究较多的是IL-17A。COPD患者诱导痰中IL-17分泌增加,增加程度与气流受限和肺功能下降相关,吸入糖皮质激素后痰中IL-17仍然增加,而中性粒细胞数没有变化[27]。(5)IL-18:以前被称作INF-γ诱导因子,于1995年首次纯化并克隆出人类的INF-γ诱导因子,后被命名为IL-18。Kang等[28]通过在转基因小鼠肺组织高表达IL-18,诱导CD4+、CD8+、CD19+和NK1.1+细胞聚集和细胞内IFN-γ、IL-17A和IL-13的水平升高,并证实IFN-γ、IL-17A和IL-13都参与了IL-18介导的COPD的发生和进展,进一步分析IL-18介导IFN-γ抑制巨噬细胞、淋巴细胞、嗜酸性粒细胞聚集,参与肺气肿和细胞毒反应;IL-18介导IL-17A和IL-13参与气道和血管重塑,推测IL-18对COPD的病理改变起中心作用。
2.2.3 趋化因子 (1)趋化因子CXCL-8:在COPD稳定期和急性加重期均发现痰及支气管肺泡灌洗液中CXCL-8水平增加,目前认为CXCL-8通过CXCR1和CXCR2受体引起中性粒细胞的趋化。有研究显示,CX CL-8被单克隆抗体中和后,中性粒细胞招募数量下降到12%的基准水平,而未被单克隆抗体中和的对照组为61.9%,且CXCL-8单克隆抗体的抑制作用呈现浓度相关性,在CXCL-8单克隆抗体浓度达到0.1 mg/mL,中性粒细胞趋化就完全降到基准水平[29]。选择性CXCR2拮抗剂和非选择性的CXCR1/CXCR2拮抗剂均能够降低CXCL-8诱导的中性粒细胞招募数量,但非选择性拮抗剂具有更强的抑制效果,且在低浓度(1 μg/mL)时只有非选择性拮抗剂有抑制作用[29]。(2)CXCL-1:Corhay等[30]研究发现,CXCL-1在健康人和COPD稳定期中没有升高,而在COPD急性加重期(呼出气冷凝物检测结果)会出现明显升高,随着病情好转,CXCL-1会明显下降,推测CXCL-1只参与COPD急性加重期的炎性反应。(3)CXCL-5:CXCL-5在不同的炎症模型中起着不同作用,有时促进炎性反应,有时抑制炎性反应。在大肠杆菌诱导的肺部炎症模型中,CXCL-5通过抑制趋化因子清除、调节趋化因子组成及减弱CXCR2亲和力来增加血液中CXCL-1和CXCL-2的浓度,减少中性粒细胞向肺组织浸润,导致肺部细菌量增多而引起死亡;而在吸入脂多糖诱导的肺部炎症模型中,CXCL-5促进了中性粒细胞向肺组织的浸润[31]。在二手烟诱导的肺部炎症模型中,CXCL-5减少肺泡灌洗液中巨噬细胞浸润数量,还观察到CXCL-5具有调节肺组织中趋化因子CXCL-2和TNF-α的合成,激活核转录因子-κB(NF-κB)和促分裂素原活化蛋白激酶、表达细胞间黏附分子-1的能力[32]。
3 小结与展望
COPD病因复杂,其具体发病机制仍不清楚,目前认为是由多个因素相互作用引起,最常见的是吸烟,其次是α1-抗胰蛋白酶缺乏、职业粉尘、有害的化学物质、空气污染、呼吸道感染等。不同病因的COPD肺部炎性反应可能会有差异,可能参与的主要炎症细胞不同或释放的主要炎症介质不同。在不同的炎症模型上也观察到,使用不同的炎症刺激诱导物(如病毒感染、烟草提取物、脂多糖等)引起肺部炎性反应中,参与的主要炎症细胞和炎症介质有明显差异。未来可以利用这些新的研究成果开发出选择性更高的药物,增加治疗有效性并减少不良反应;筛选出更加有意义的COPD炎症生物标记物,更好地评价COPD严重性和预后。
[1]Fabbri LM,Hurd SS,GOLD Scientific Committee.Global strategy for the diagnosis,manage ment and prevention of COPD:2003 update[J].Eur Respir J,2003,22(1):1-2.
[2]Xia J,Zhao J,Shang J,et al.Increased IL-33 expression in chronic obstructive pulmonary disease[J].Am J Physiol Lung Cell Mol Physiol,2015,308(7):619-627.
[3]Yasuo M,Mizuno S,Allegood J,et al.Fenretinide causes emphysema, which is prevented by sphingosine 1-phoshate[J].PLoS One,2013,8(1):e53927.
[4]Barnes PJ.Alveolar macrophages as orchestrators of COPD[J].COPD,2004,1(1):59-70.
[5]Barnes PJ.Increased levels of interleukin-8 in BAL fluid from smokers susceptible to pulmonary emphysema[J].Clin Chest Med,2014,35(1):71-86.
[6]Tanino M,Betsuyaku T,Takeyabu K,et al.Increased levels of interleukin-8 in BAL fluid from smokers susceptible to pulmonary emphysema[J]. Thorax,2002,57(5):405-411.
[7]Forsslund H,Mikko M,Karimi R,et al.Distribution of T-cell subsets in BAL fluid of patients with mild to moderate COPD depends on current smoking status and not airway obstruction[J].Chest,2014,145(4):711-722.
[8]Hodge S,Hodge G,Holmes M,et al.Increased CD8 T-cell granzyme B in COPD is suppressed by treatment with low-dose azithromycin[J].Respirology,2015,20(1):95-100.
[9]Hodge G,Mukaro V,Reynolds PN,et al.Role of increased CD8/CD28(null)T cells and alternative co-stimulatory molecules in chronic obstructive pulmonary disease[J].Clin Exp Immunol,2011,166(1):94-102.
[10]Di Stefano A,Caramori G,Capelli A,et al.STAT4 activation in smokers and patients with chronic obstructive pulmonary disease[J].Eur Respir J,2004,24(1):78-85.
[11]Eltboli O,Brightling CE.Eosinophils as diagnostic tools in chronic lung disease[J].Expert Rev Respir Med,2013,7(1):33-42.
[12]Eltboli O,Mistry V,Barker B,et al.Relationship between blood and bronchial submucosal eosinophilia and reticular basement membrane thickening in chronic obstructive pulmonary disease[J].Respirology,2015,20(4):667-670.
[13]Barnes PJ.Mediators of chronic obstructive pulmonary disease[J].Pharmacol Rev,2004,56(4):515-548.
[14]Silva MT.Neutrophils and macrophages work in concert as inducers and effectors of adaptive immunity against extracellular and intracellular microbial pathogens[J].J Leukoc Biol,2010,87(5):805-813.
[15]Sadik CD,Kim ND,Iwakura Y,et al.Neutrophils orchestrate their own recruitment in murine arthritis through C5aR and FcγR signaling[J].Proc Natl Acad Sci USA,2012,109(46):3177-3185.
[16]Afonso PV,Janka-Junttila M,Lee YJ,et al.LTB4 is a signal-relay molecule during neutrophil chemotaxis[J].Dev Cell,2012,22(5):1079-1091.
[17]Drozdovszky O,Barta I,Antus B.Sputum eicosanoid profiling in exacerbations of chronic obstructive pulmonary disease[J].Respiration,2014,87(5):408-415.
[18]Dagouassat M,Gagliolo JM,Chrusciel S,et al.The cyclooxygenase-2-prostaglandin E2 pathway maintains senescence of chronic obstructive pulmonary disease fibroblasts[J].Am J Respir Crit Care Med,2013,187(7):703-714.
[19]Chen Y,Chen P,Hanaoka M,et al.Enhanced levels of prostaglandin E2 and matrix metalloproteinase-2 correlate with the severity of airflow limitation in stable COPD[J].Respirology,2008,13(7):1014-1021.
[20]Makwana R,Gozzard N,Spina D,et al.TNF-α-induces airway hyperresponsiveness to cholinergic stimulation in guinea pig airways[J].Br J Pharmacol,2012,165(6):1978-1991.
[21]Botelho FM,Bauer CM,Finch D,et al.IL-1α/IL-1R1 expression in chronic obstructive pulmonary disease and mechanistic relevance to smokeinduced neutrophilia in mice[J].PLoS One,2011,6(12):e28457.
[22]Pauwels NS,Bracke KR,Dupont LL,et al.Role of IL-1α and the Nlrp3/ caspase-1/IL-1β axis in cigarette smoke-induced pulmonary inflammation and COPD[J].Eur Respir J,2011,38(5):1019-1028.
[23]Sichelstiel A,Yadava K,Trompette A,et al.Targeting IL-1β and IL-17A driven inflammation during influenza-induced exacerbations of chroniclung inflammation[J].PLoS One,2014,9(2):e98440.
[24]Hackett TL,Stefanowicz D,Aminuddin F,et al.Effect of gene environment interactions on lung function and cardiovascular disease in COPD[J].Int J Chron Obstructi Pulmon Dis,2011,6:277-287.
[25]He JQ,Foreman MG,Shumansky K,et al.Associations of IL6 polymorphisms with lung function decline and COPD[J].Thorax,2009,64(8):698-704.
[26]Celli BR,Locantore N,Yates J,et al.Inflammatory biomarkers improve clinical prediction of mortality in chronic obstructive pulmonary disease[J]. Am J Respir Crit Care Med,2012,185(10):1065-1072.
[27]Doe C,Bafadhel M,Siddiqui S,et al.Expression of the T helper 17-associated cytokines IL-17A and IL-17F in asthma and COPD[J].Chest,2010,138(5):1140-1147.
[28]Kang MJ,Choi JM,Kim BH,et al.IL-18 induces emphysema and airway and vascular remodeling via IFN-γ,IL-17A,and IL-13[J].Am J Respir Crit Care Med,2012,185(11):1205-1217.
[29]Kaur M,Singh D.Neutrophil chemotaxis caused by chronic obstructive pulmonary disease alveolar macrophages:the role of CXCL8 and the receptors CXCR1/CXCR2[J].J Pharmacol Exp Ther,2013,347(1):173-80.
[30]Corhay JL,Moermans C,Henket M,et al.Increased of exhaled breath condensate neutrophil chemotaxis in acute exacerbation of COPD[J].Respir Res,2014,15:115.
[31]Balamayooran G,Batra S,Cai S,et al.Role of CXCL5 in leukocyte recruitment to the lungs during secondhand smoke exposure[J].Am J Respir Cell Mol Biol,2012,47(1):104-111.
[32]Mei J,Liu Y,Dai N,et al.CXCL5 regulates chemokine scavenging and pulmonary host defense to bacterial infection[J].Immunity,2010,33(1):106-117.
10.3969/j.issn.1009-5519.2015.18.022
A
1009-5519(2015)18-2787-04
2015-04-18)
周复(1980-),女,重庆巴南人,主治医师,主要从事呼吸内科临床工作;E-mail:zhouf2015@sina.com。
罗治海(E-mail:1271301155@qq.com)。
