基于多视角特征融合与随机森林的蛋白质结晶预测
2015-07-09李强等
李强等


摘 要: X射线晶体结构分析是测定蛋白质结构的重要方法之一,国际蛋白质数据库(PDB)中已知晶体结构的蛋白质80%~90%均是使用该方法得到的。然而,并不是所有的蛋白质都能良好结晶,使用晶体结构分析方法对不能结晶的蛋白质进行结构测定将浪费大量的资源。因此,研发准确高效的算法来对蛋白质能否结晶进行预测就具有重要意义。在此提出了一种组合蛋白质物理化学特性、序列信息与进化信息的蛋白质结晶预测方法。该方法从不同视角抽取分别抽取蛋白质的物理化学特征、伪氨基酸组成特征(PseAAC)和伪位置特异性得分矩阵特征(PsePSSM),使用随机森林对组合的特征进行蛋白质结晶预测。在标准数据集上的独立测试验证的结果表明,这里所述的蛋白质结晶预测方法具有良好的性能。
关键词: 蛋白质结晶; 伪氨基酸组成; 位置特异性得分矩阵; 随机森林
中图分类号: TN911?34 文献标识码: A 文章编号: 1004?373X(2015)08?0050?04
Protein crystallization prediction based on fusion of multi?view features
and random forest
LI Qiang1, ZHENG Yu?jie2
(1. School of Computer Science and Engineering, NUST, Nanjing 210094, China;
2. The 28th Research Institute, China Electronics Technology Group Corporation, Nanjing 210007, China)
Abstract: The X?ray crystallography analysis is one of the important methods to measure protein structure, by which 80%~90% protein of the known crystalloid structures in the international protein data bank (PDB) has been obtained. However, not all the proteins used for determining structures are crystallizable, which will lead to a low success rate of crystallization projects and a serious waste of resources to measure those non?crystallizable protein. Hence, it is important to develop an accurate and effective method for predicting whether a protein will crystallize. In this study, a new protein crystallization prediction method to combine the protein physicochemical characteristic, serial information and evolutionary information is proposed, which extracts the protein physicochemical properties, pseudo amino acids composition (PseAAC) and pseudo position specific scoring matrix (PsePSSM) at different visual angle respectively. The random forest is taken as classifier predict protein crystallization of the combined Properties. Experimental results on benchmark dataset over cross?validation test and independent validation test show that the proposed method has perfect performance.
Keywords: protein crystallization; pseudo amino acids composition; position specific scoring matrix; random forest
蛋白质的功能特性与它的三维结构密切相关。准确获取蛋白质的三维结构信息对于理解蛋白质的功能以及蛋白质与其他生物小分子的相互作用至关重要。X射线晶体结构分析、核磁共振光谱法(NMR)以及电子显微镜是测定蛋白质三维结构的常用方法。然而,并不是所有的蛋白质都能良好结晶,使用X射线晶体结构分析方法对不能结晶的蛋白质进行结构测定将浪费大量的资源。因此,研发准确高效的算法来对蛋白质能否结晶进行预测就具有重要意义。
目前,研究人员已经研发出很多预测蛋白质结晶的有效方法,如:OB?score,CRYSTALP,XtalPred,ParCrys,MetaPPCP,CRYSTALP2,MCSG?Z score,PCCpred,以及RFCRYS等。分析这些方法可以发现:
(1) 氨基酸组成成分(Amino Acids Composition, AAC)以及伪氨基酸组成成分(Pseudo Amino Acids Composition,PseAAC)是常用的特征表示方法;此外,氨基酸的物理化学属性以及通过预测方法获得的蛋白质结构属性也往往被用于蛋白质的特征表示;
(2) 蛋白质的进化信息是一种有效的特征表示方法,并且已经被广泛地应用于很多蛋白质属性预测,但是,蛋白质结晶预测方法中没有一个使用蛋白质的进化信息来进行特征表示;
(3) 虽然现有方法在蛋白质结晶预测问题上取得了重要进展,但是预测性能还有进一步提高的空间。
基于上述分析,本文首先考察蛋白质进化信息能否用于蛋白质结晶预测问题的特征表示。然后,将蛋白质物理化学信息、序列信息及进化信息进行组合用于蛋白质结晶预测,以进一步提高预测性能。在标准数据集上的交叉验证及独立测试验证的结果表明,本文所述的方法具有良好的性能,是对现有蛋白质结晶预测方法的有益补充。
1 数据来源
数据集S表示为:
[S=S+?S-] (1)
式中:S+表示正样本集,其中包含的是能结晶的蛋白质序列; S-表示负样本集,其中包含的是不能结晶的蛋白质序列;符号[?]表示集合理论中的并集。本文中使用Kurgan等构建的数据集[1],该数据集包含一个训练子集(Train1500)和一个独立测试子集(Test500)。Train1500中包含756个正样本和744个负样本,Test500中包含244个正样本和256个负样本。为了进一步验证本文所述方法的泛化能力,还使用了Overton等人构建的另外一个独立测试集Test144,其中包含72个正样本和72个负样本[2]。在数据集的构建过程,已经考虑了蛋白质之间的同源冗余性消除,蛋白质序列之间的同源性[1]小于25%。
2 多视角特征提取
2.1 物理化学特征
蛋白质的一些物理化学性质对蛋白质能否结晶有着重要影响。因此,本文依据氨基酸属性集AAIndex1,筛选出7个物理化学性质:疏水性指数、平均极性、正电荷、负电荷、净电荷、等电位和分子质量。每条蛋白质的上述7个物理化学性质构成一个维数为7的特征向量。
2.2 伪氨基酸组成成分特征提取
PseAAC是由Chou在经典的AAC特征基础上提出来的,分为I型和Ⅱ型。一个蛋白质的Ⅱ型PseAAC特征向量可表示为[20+iλ](其中[i]表示生成PseAAC时使用的氨基酸属性的数量,[λ]表示序列相关因子)。PseAAC生成方法如下:
4 实验结果和讨论
4.1 独立测试验证结果
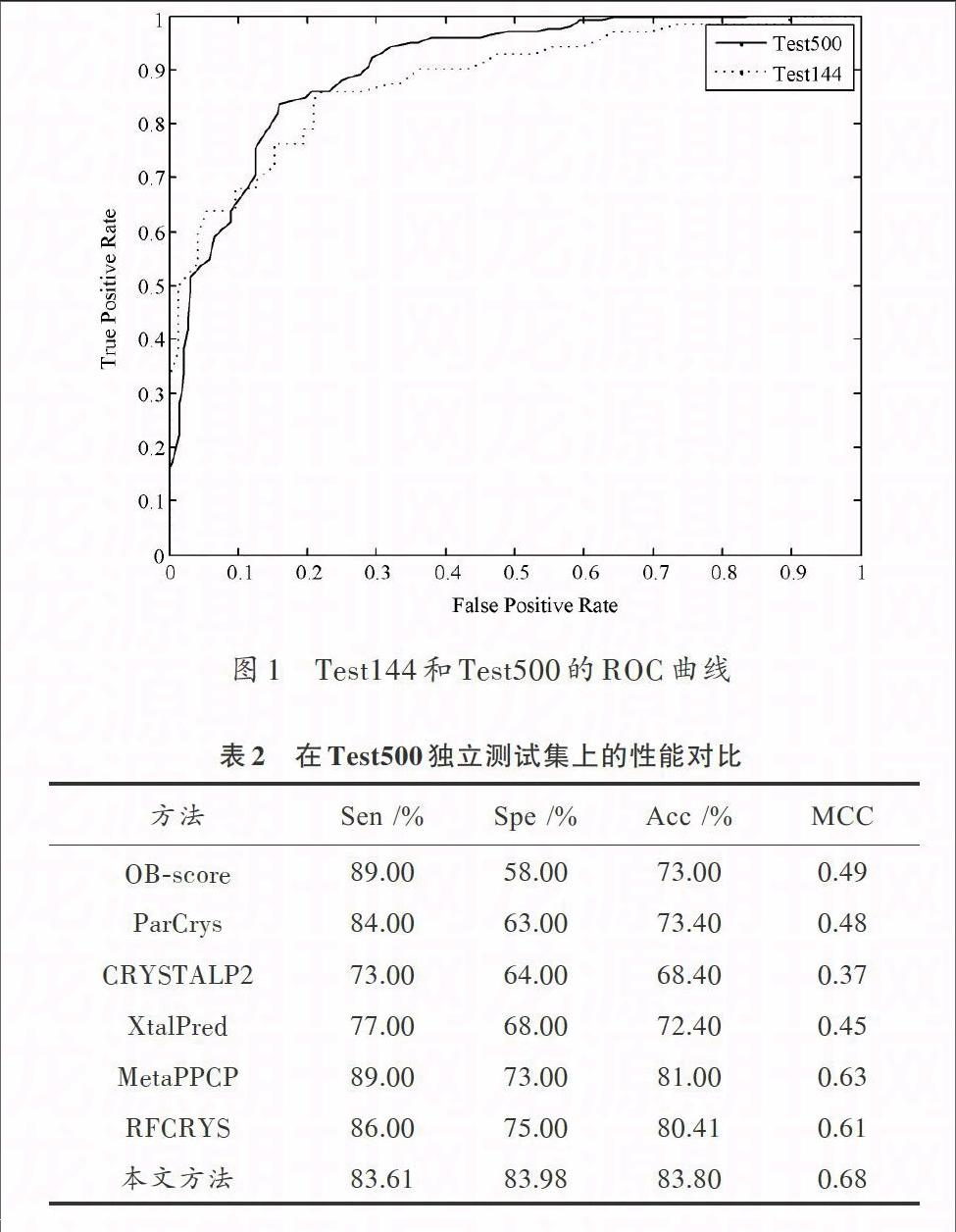
表1和表2分别列出了本文方法与其他蛋白质结晶预测方法在独立测试集Test144及Test500上的性能对比[3]。图1给出了本文方法在独立测试集Test144和Test500上的ROC曲线。
从表1可以看出,在Test144独立测试上,本文所述方法的亦获得了较好的性能,Acc以及MCC分别为81.94%和0.64,取得了和RFCRYS相当的预测性能。另一方面,虽然OB?score的Sen达到了88.00%,但是其Spe仅仅为47.00%,表明OB?score方法的预测结果中存在大量的假阳性(FP)。
表2 在Test500独立测试集上的性能对比
从表2的结果来看,本文所述方法在独立测试集Test500上再次取得了最好的性能。Spe、Acc以及MCC分别为83.98%,83.80%和0.68,比RFCRYS分别高出了约9%,2%以及7%。结合表1及表2的结果,可以看出本文所述的方法较之于已有的蛋白质结晶预测方法有着更为良好的泛化能力。这得益于使用了多种有效的蛋白质特征以及强有力的随机森林分类算法。
5 结 语
本文提出一种组合蛋白质物理化学特征、伪氨基酸组成特征以及伪位置特异性得分矩阵特征的蛋白质结晶预测方法。该方法同时利用了蛋白质的序列及进化信息,因此所抽取的特征更具有鉴别能力。在标准数据集上独立测试验证结果表明,本文所述的方法具有良好的性能,是对已有蛋白质结晶预测方法的有益补充。
参考文献
[1] KURGAN L, RAZIB A A, AGHAKHANI S, et al. CRYSTALP2: sequence?based protein crystallization propensity prediction [J]. BMC Structural Biology, 2009, 9: 50?63.
[2] OVERTON I M, PADOVANI G, GIROLAMI M A, et al. ParCrys: a Parzen window density estimation approach to protein crystallization propensity prediction [J]. Bioinformatics, 2008, 24(7): 901?907.
[3] JAHANDIDEH S, MAHDAVI A. RFCRYS: Sequence?based protein crystallization propensity prediction by means of random forest [J]. Journal of Theoretical Biology, 2012, 306: 115?119.
[4] YU D J, HU J, WU X W, et al. Learning protein multi?view features in complex space [J]. Amino Acids, 2013, 44(5): 1365?1379.
[5] 王建,王彩芸.基于改进牛顿算法的蛋白质二级结构预测[J].现代电子技术,2009,32(14):135?137.
[6] 李秀娟,田川,冯欣.数据挖掘分类技术研究与分析[J].现代电子技术,2010,33(20):86?88.
