肝螺杆菌对肝癌细胞侵袭能力的影响及其可能分子机制
2015-05-07张春忙彭凡洲邓慧敏
张春忙 彭凡洲 邓慧敏 王 菲 刘 江 白 杨
1994年,在国际癌症研究中心(International A-gency for Research on Cancer,IARC)公布的人类致癌物名单中,幽门螺杆菌被确定为人类Ⅰ类致癌因子[1]。人类首次将细菌感染与癌症的发生联系在一起。我国是肝癌高发区,在肝癌的病因研究方面,目前认为HBV和HCV感染是原发性肝癌最常见的病因,其次还与黄曲霉素、酒精、自身免疫等因素有关[2]。大量临床研究表明,尽管存在的致癌因素相似,但是其临床表现及预后却相差甚远。因此,疾病表现除了与致癌因素和宿主免疫状况密切相关外,可能还存在其他协同致病因素,如细菌感染等[3],近年来这逐渐成为人们关注的焦点[4]。大量动物实验研究已证实,肝螺杆菌(Helicobacter Hepaticus,H.hepaticus)感染与啮齿类动物肝癌的发生有显著的相关性[5-7],国内外学者在人类肝癌组织中发现H.hepaticus 16sRNA也有不少报道[8],但目前针对H.hepaticus与肝癌细胞的直接作用及其可能机制研究相对较少。因此,在离体条件下,研究H.hepaticus能否作用于肝癌细胞,这将有助于进一步认识H.hepaticus感染与肝癌发生发展的关系。
本研究旨在探讨H.hepaticus对肝脏肿瘤细胞本身的直接作用及H.hepaticus活菌对肝癌细胞侵袭能力的影响及其可能机制。
材料和方法
一、实验材料
肝癌细胞株HEPG2和HCCLM3,南方医院消化内科实验室冻存。H.hepaticus标准菌株ATCC51450,购自美国国家标准菌种保藏中心(American Type Culture Collection,ATCC)。南美胎牛血清(FBS),购自美国Hyclone公司。DMEM高糖培养基,均购自美国Gibco公司。兔抗人TLR4和MyD88、TRIF多克隆抗体,购自美国Abcam公司。鼠抗人GAPDH单克隆抗体、辣根过氧化酶标记的山羊抗鼠(兔)二抗,购自北京中杉金桥生物公司。
二、方法
1.细菌培养
将冻存的H.hepaticus标准菌株于含8%灭活脱纤维羊血的布氏琼脂培养基复苏,37℃、微需氧(5%O2,10%CO2,85%N2)、高湿度条件下培养 5 ~ 7 d,根据菌落性状、形态学特征及生化反应鉴定为H.hepaticus后,纯培养2~3代,应用PBS收集细菌,4℃3 000 rpm离心10 min,弃上清,应用PBS重新悬浮,于分光光度计上调节H.hepaticus浓度为1×109CFU/mL备用,并将H.hepaticus菌液连续3次作10倍稀释(1×108CFU/mL、1×107CFU/mL和1×106CFU/mL)。
2.细胞与细菌共孵育
将人肝癌细胞株HEPG2和HCCLM3接种于盛有双抗的含10%胎牛血清的DMEM培养基的培养瓶,置37℃、含5%CO2、饱和湿度的环境中培养,每隔2 d弃去旧液,换以新鲜培养基,待培养至细胞融合为单层后,用0.25%胰蛋白酶消化,制成单细胞悬液,按预试验结果配制成1×106/mL细胞浓度,以每孔1 mL接种于6孔培养板中,放入细胞培养箱中培养。细胞贴壁后,吸去原培养液,PBS洗涤,更换无血清的DMEM培养基。实验组加入1×109CFU/mL、1×108CFU/mL、1×107CFU/mL 和 1×106CFU/mL四个不同浓度的H.hepaticus活菌刺激肝癌细胞,每组分别取12 h、24 h、48 h、72 h 4个时间点。对照组仅加入无H.hepaticus的空白液体培养基。
3.体外侵袭试验
将H.hepaticus处理的实验组细胞用PBS洗涤2次,0.25%胰酶消化细胞,l 100 rpm离心5 min,弃上清,DMEM培养基(含1%FBS)重悬细胞,细胞计数,调整HEPG2及HCCLM3细胞密度至1×106/mL。Transwell小室下室加入500 μL含10%FBS的DMEM培养基作为趋化因子,接种细胞,细胞悬液200 μL加入Transwell小室上室,然后盖好盖子,置于37℃、5%CO2饱和湿度的细胞培养箱中培养24 h。用棉签擦去基质胶和上室内的细胞,通过给细胞0.1%结晶紫室温染色20 min,可在光学纤维镜下计数细胞,选择中及上、下、左、右共5个象限计数。
4.蛋白质免疫印迹分析(Western blot)
用1×108CFU/mL的H.hepaticus刺激HEPG2和HCCLM3细胞48 h后,采用RIPA裂解液提取细胞蛋白,4℃13 000 g离心20 min后,收集上清液,BCA法进行蛋白含量测定。用移液枪吸取所需上样样品至EP管中,加入5×loading buffer稀释至1×的终浓度。上样前要将样品加热5 min使蛋白变性,将40 μg该蛋白质加载到10%的SDS-PAGE中电泳,经浓缩胶(80 V、30 min),分离胶(120 V、90 min)分离后,电转移到硝酸纤维素膜上。将硝酸纤维素膜放在含有5%脱脂奶粉封闭液中,置于摇床上室温孵育1 h后用TBST缓冲液洗膜,分别加入相应的封闭液稀释的一抗(兔抗人TLR4,MyD88及TRIF多克隆抗体和鼠抗人GAPDH单克隆抗体(稀释浓度均为1:1 000)),置于4℃摇床孵育过夜,再用0.01 mol/L TBST缓冲液洗膜,加入合适的酶标二抗 (辣根过氧化物酶偶联的二抗溶液山羊抗兔IgG和山羊抗鼠IgG),TBST洗膜,置于室温下摇床缓慢振荡。化学发光剂(ECL)检测转印膜上靶蛋白信号。
5.荧光定量 PCR(real time PCR)
用1×108CFU/mL的H.hepaticus刺激HEPG2和HCCLM3细胞48 h后,采用RNAiso Plus提取细胞的总RNA,紫外分光光度计测定RNA浓度。取2 μg RNA作为模板,根据Strand cDNA Synthesis Kit说明书反转录为cDNA,取cDNA进行PCR扩增,根据SYBRPremix Ex Taq II说明书进行Real time PCR,25 μL的PCR反应体系。PCR程序:95℃30 s,预变性;95 ℃ 5 s,60 ℃ 30 s,共 40 个循环。本研究中所使用的引物由上海生工生物工程公司合成(表 1)。

表1 检测基因引物序列
6.RNA干扰
根据siRNA oligomer和lipofectamineTM 2000说明书,建立siRNA干扰系统,诱导TLR4、MYD88基因沉默,下调HEPG2和HCCLM3细胞中TLR4和MyD88基因的表达,使用Western Blot检测TLR4、MyD88表达水平,验证siRNA干扰的效果。分别设置TLR4-siRNA、MyD88-siRNA干扰组、阴性对照组(转染无关序列)和空白对照组(空白转染),用H.hepaticus作用为各组细胞,通过Transwell实验检测各组细胞侵袭能力是否有变化。小干扰RNA(siRNA)的寡核苷酸序列(阴性对照ctrlsiRNA:5′-UUC UCC GAA CGU GUC ACG U-3′;MyD88-siRNA:5′-AAG GCA AUG AAG AAA GAG UU C-3;TLR4-siRNA:5′-AAC TTG TAT TCA AGG TCT GGC-3′)。
三、统计学处理
结 果
一、H.hepaticus增加人肝癌细胞的侵袭能力
通过Transwell实验,用 1× 109CFU/mL、1×108CFU/mL、1×107CFU/mL、1×106CFU/mL不同浓度的H.hepaticus刺激肝癌细胞后,在12 h、24 h、48 h、72 h时间点检测侵袭细胞数量,以此观察浓度不同、刺激时间不同情况下H.hepaticus对肝癌细胞的侵袭能力的影响。并依此为根据,确定后续实验的最佳刺激浓度及刺激时间。实验结果表明,如图1所示,随着H.hepaticus活菌刺激浓度的增加,穿过基质膜的侵袭性细胞的数量明显增加,且在1×108CFU/mL达到峰值。如图2所示,我们发现相同浓度刺激时,随着时间的延长,穿过基质膜的侵袭性细胞的数量明显增加,且在刺激48 h时达到峰值。基于该实验结果,我们后续实验H.hepaticus刺激浓度选为1×108CFU/mL,刺激时间选为48 h。我们的数据显示H.hepaticus可促进肝癌细胞的侵袭能力,且呈时间及浓度依懒性,在一定范围内,随着H.hepaticus刺激浓度及时间的延长,促进肝癌细胞的侵袭能力越强。

图1 HEPG2或HCCLM3细胞经不同浓度H.hepaticus刺激

图2 HEPG2或HCCLM3经不同时间的H.hepaticus刺激
二、H.hepaticus激活肝癌细胞内的 TLR4/MyD88信号通路
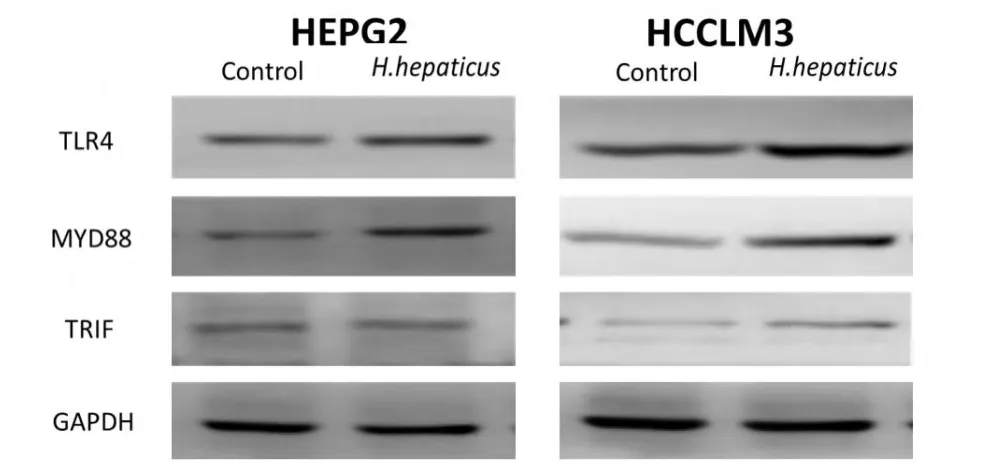
图3 HEPG2和HCCLM3细胞经或不经H.hepaticus剌激TLR4、MyD88、TRIF 蛋白表达水平
我们用Western Blot的方法检测经过H.hepaticus刺激48 h后,人肝癌细胞HEPG2和HCCLM3细胞中TLR4、MyD88和TRIF的表达情况。如图3,结果显示,经H.hepaticus刺激后,肝癌细胞HEPG2和HCCLM3中TLR4、MyD88蛋白水平明显上调(P<0.05),但对TRIF的表达没有明显影响 (P>0.05)。我们用利Real time PCR的方法对此进行了进一步的验证,如图4,mRNA水平也证明了相似的结果。结果说明,H.hepaticus可能通过MyD88介导TLR4的信号通路转导来激活TLR4信号通路,而不是通过TRIF介导。

图4 HEPG2和HCCLM3细胞经或不经H.hepaticus剌激TLR4、MyD88、TRIF 基因表达水平(*P<0.05;**P>0.05)
三、H.hepaticus通过影响TLR4/MyD88信号通路增强人肝癌细胞的侵袭能力
为进一步证实TLR4/MyD88信号通路与H.hepaticus增强人肝癌细胞侵袭能力的关系,我们通过小RNA干扰方法下调肝癌细胞HEPG2和HCCLM3中TLR4和MyD88的蛋白表达水平(图5),经H.hepaticus刺激后用Transwell实验检测各组肝癌细胞的侵袭能力。如图6,与正常对照组相比,TLR4或MyD88基因沉默后,H.hepaticus刺激不能提高肝癌细胞的侵袭能力,此时肝癌细胞的侵袭能力下降(P<0.05),结果说明H.hepaticus增强人肝癌细胞的侵袭能力是依赖TLR4/MyD88信号通路而实现的。
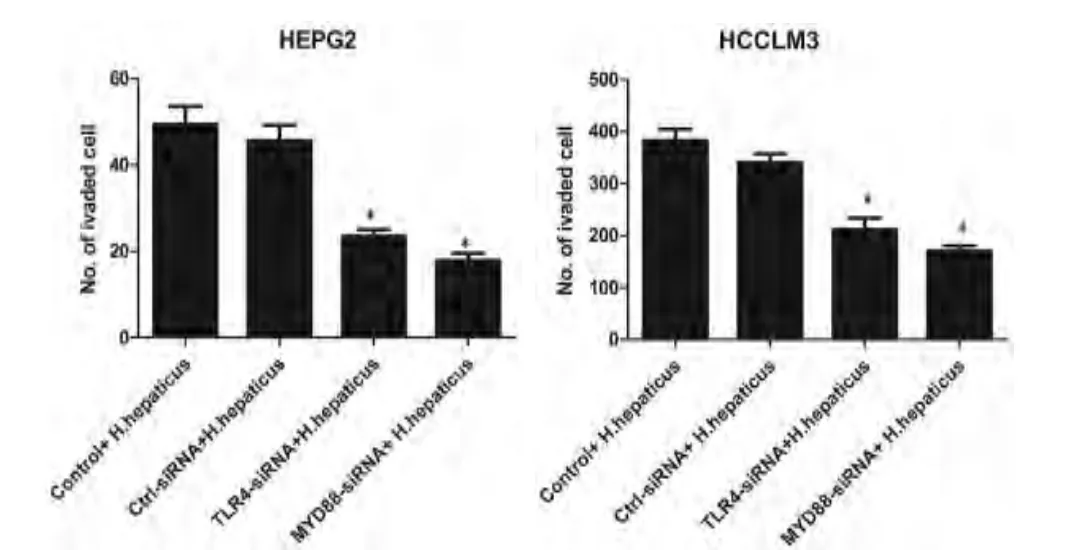
图6 HEPG2和HCCLM3细胞转染了靶向TLR4-siRNA、MyD88-siRNA和对照siRNA后,经H.hepaticus刺激,siRNA下调TLR4、MyD88表达水平后的细胞的侵袭能力明显下降,Control为未转染对照,*P<0.05。
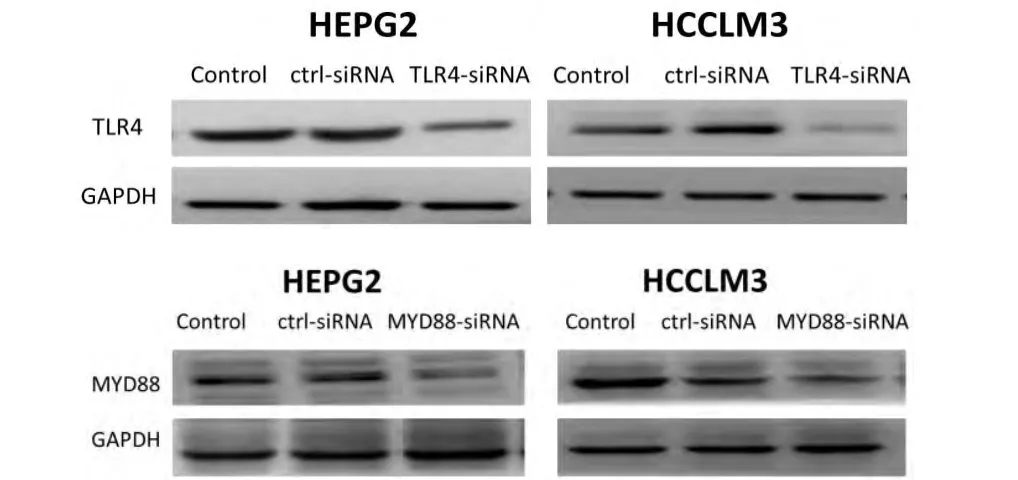
图5 HEPG2和HCCLM3细胞转染了靶向TLR4-siRNA、MyD88 siRNA和对照siRNA后,TLR4、MyD88的蛋白表达水平(Control为未转染对照)
讨 论
流行病学研究表明,螺杆菌感染与肝癌发病率呈正相关,这提示螺杆菌可能在肝癌的发生发展中具有重要作用[2]。大量动物实验资料显示,H.hepaticus感染和肝脏炎症及肿瘤的发生具有明显相关性,国内外许多学者利用PCR、DNA序列分析证实了人肝癌组织中确实存在H.hepaticus感染,但至今细菌培养技术仍未成功从人体标本中分离出活菌,H.hepaticus是否具有致癌作用仍难以证明[8]。本课题组前期研究显示,感染HBV和HVC的肝癌患者血清抗H.hepaticus IgG浓度与病毒DNA量及AFP浓度呈正比,提出了H.hepaticus感染可能作为单独或辅助因素促进肝脏肿瘤发生发展。以往研究H.hepaticus主要集中在动物实验及组织水平探讨上,H.hepaticus对肝脏肿瘤细胞本身的影响鲜有报道。本研究旨在阐述H.hepaticus对肿瘤细胞本身的直接作用,研究H.hepaticus活菌对肝癌细胞侵袭能力的影响。
早期肝癌患者并没有特异性的临床症状及体征,因此临床上就诊的肝癌患者大多数已发展为晚期肝癌,甚至出现远处转移,通常已经错过最佳的手术时机。肝癌患者预后的影响因素很多,其中最重要的两个原因为侵袭和转移。肿瘤导致患者死亡的主要原因之一,是在肿瘤发生发展过程中出现的远处转移及其引发的相关并发症。深入研究肝脏肿瘤的浸润及转移机制,将有助于开发针对浸润及转移的分子的基因靶向治疗,具有重要的临床意义[9-11]。
Toll样受体是一类天然免疫受体,属于Ⅰ型跨膜蛋白受体,最早在果蝇中发现。此类受体与固有免疫密切相关,能够识别病原体相关分子模式(pathogen-associated molecular patterns,PAMPs),是近年来发现的一类重要的模式识别受体(pattern recognitionreceptor,PRR)。随着对TLRs深一步的研究,发现TLRs的分布十分广泛,主要表达于天然免疫细胞,如单核细胞、巨噬细胞、树突状细胞、多形核细胞、淋巴细胞及自然杀伤细胞等表面,它能激活免疫细胞分泌大量炎性细胞因子和趋化因子,在发挥机体抗病原微生物感染能力中起关键作用[12]。TLRs在很多肿瘤细胞及组织中都有功能性表达,大量研究认为TLRs参与肿瘤细胞的生物学行为改变,并在肿瘤发生发展过程中起着重要作用[13],特别是与炎症相关肿瘤,如肝癌、结肠癌、胃癌等[14-16]。TLRs是进化过程中比较保守的一个受体家族,至今包括13个成员陆续被发现,它在天然免疫和获得性免疫中都承担着相当重要的角色,是连接天然免疫和获得性免疫的桥梁。
TLR4是人类发现的第一个Toll样受体,几乎分布于所有的细胞系。最初人们认为TLR4主要表达于免疫细胞特别是巨噬细胞和树突状细胞表面,一旦识别并结合PAMPs,将激活天然免疫细胞,就会导致下游信号事件和转录因子的激活,最终诱导分泌一系列抗微生物分子、化学趋化因子、细胞因子和共刺激分子,参与机体的抗感染免疫。研究证实,在TLR4信号通路中,存在MyD88依赖途径和TRIF依赖途径的2条信号通路。在MyD88依赖的通路中,MyD88依赖途径需要由MyD88承接转导,是几乎所有TLRs信号转导的共同通路 (TRL3除外),而TRIF依赖途径,即是MyD88非依赖途径,由β干扰素TIR结构域衔接蛋白 (TIR-domaincontaining adaptor inducinginterferon-β,TRIF)联接,主要存在于TLR3和TLR4通路中。近年来发现,TLR4也广泛表达于在多种肿瘤细胞和组织中,并与肿瘤的分化程度及预后密切相关(如肺癌、卵巢癌、胰腺癌及肝癌等)[17-20]。目前在对消化系统疾病研究过程中,研究发现TLR4在一些慢性肝脏疾病如病毒性肝炎、肝硬化、肝纤维化、肝癌及酒精性肝病的发生发展过程中具有重要作用[21]。
在本研究中,我们首先用Transwell侵袭实验,发现肝癌细胞HEPG2和HCCLM3在不同浓度、不同时间接受H.hepaticus刺激后,侵袭细胞数量显著增加,且呈时间及浓度依懒性,且在1×108CFU/mL刺激48 h到达峰值。为了探讨H.hepaticus提高肝癌细胞侵袭能力的可能分子机制,我们检测H.hepaticus刺激是否可激活肝癌细胞中的TLR4信号,结果表明,经处理后HEPG2和HCCLM3细胞TLR4蛋白和mRNA表达水平明显升高,且H.hepaticus可有效上调TLR4信号接头蛋白MyD88的表达,却对TRIF的表达没有明显影响。这些结果提示,H.hepaticus可有效活化肝细胞癌中的TLR4/MyD88信号通路。我们后续进一步研究H.hepaticus促进肝癌细胞侵袭能力的增强是否由TLR4/MyD88信号所介导,我们采用siRNA技术下调肝癌细胞中TLR4和MyD88的表达水平,结果显示,将HEPG2细胞和HCCLM3细胞分别转染TLR4和MyD88的siRNA后,可显著下调其TLR4和MyD88蛋白水平的表达。siRNA转染能够有效抑制H.hepaticus对HEPG2细胞和HCCLM3侵袭能力的增强作用,说明TLR4/MyD88信号在H.hepaticus促进肝细胞癌的侵袭中具有重要作用。
H.hepaticus感染与肝癌的发生发展具有重要的研究价值,尤其是对H.hepaticus的致病机制以及对肝癌病原学方面的病因的探索具有重要意义。目前对于H.hepaticus致病机制的研究尚处于初级阶段,H.hepaticus对肝癌细胞侵袭力及其他生物学功能的影响有待进一步深入研究,H.hepaticus对肝癌细胞TLRs信号通路及其他相关通路的影响也有待进一步阐明。
1 IARC working group on the evaluation of carcinogenic risks to humans:some industrial chemicals.Lyon,15-22 February 1994.IARC Monogr Eval Carcinog Risks Hum,1994,60:1-560.
2 Pellicano R,Menard A,Rizzetto M,et al.Helicobacter species and liver diseases:association or causation?Lancet Infect Dis,2008,8(4):254-260.
3 Krüttgen A,Horz H,Weber-Heynemann J,et al.Study on the association of helicobacter species with viral hepatitis-induced hepatocellular carcinoma.Gut Microbes,2014,3(3):228-233.
4 Francescone R,Hou V,Grivennikov SI.Microbiome,inflammation,and cancer.Cancer journal(Sudbury,Mass.),2014,20(3):181-189.
5 Rogers AB,Boutin SR,Whary M T,et al.Progression of chronic hepatitis and preneoplasia in Helicobacter hepaticus-infected A/JCr mice.Toxicol Pathol,2004,32(6):668-677.
6 Fox JG,Li X,Yan L,et al.Chronic proliferative hepatitis in A/JCr mice associated with persistent Helicobacter hepaticus infection:a model of helicobacter-induced carcinogenesis.Infect Immun,1996,64(5):1548-1558.
7 Ward JM,Fox JG,Anver MR,et al.Chronic active hepatitis and associated liver tumors in mice caused by a persistent bacterial infection with a novel Helicobacter species.J Natl Cancer Inst,1994,86(16):1222-1227.
8 Yang J,Ji S,Zhang Y,et al.Helicobacter hepaticus infection in primary hepatocellular carcinoma tissue.Singapore Medical Journal,2013,54(8):451-457.
9 Han Y,Zhang Y,Jia T,et al.Molecular mechanism underlying the tumor-promoting functions of carcinoma-associated fibroblasts.Tumour Biol,2015,36(3):1385-1394.
10 Bruix J,Boix L,Sala M,et al.Focus on hepatocellular carcinoma.Cancer Cell,2004,5(3):215-219.
11 Chew V,Tow C,Teo M,et al.Inflammatory tumour microenvironment is associated with superior survival in hepatocellular carcinoma patients.J Hepatol,2010,52(3):370-379.
12 McClure R,Massari P.TLR-Dependent Human Mucosal Epithelial Cell Responses to Microbial Pathogens.Front Immunol,2014,5:386.13 Machida K.TLRs,Alcohol,HCV,and Tumorigenesis.Gastroenterology Research and Practice,2010,2010:1-8.
14 Aoyama T,Paik Y,Seki E.Toll-Like Receptor Signaling and Liver Fibrosis.Gastroenterology Research and Practice,2010,2010:22-30.15 Fukata M,Chen A,Vamadevan AS,et al.Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors.Gastroenterology,2007,133(6):1869-1881.
16 Casta魨 O-Rodr魨 Guez N,Kaakoush NO,Mitchell HM.Pattern-Recognition Receptors and Gastric Cancer.Front Immunol,2014,5:336.
17 Ahmed A,Redmond HP,Wang JH.Links between Toll-like receptor 4 and breast cancer.Onco Immunol,2014,2(2):e22945.
18 Wang L,Zhu R,Huang Z,et al.Lipopolysaccharide-Induced Toll-Like Receptor 4 Signaling in Cancer Cells Promotes Cell Survival and Proliferation in Hepatocellular Carcinoma.Digest Dis Sci,2013,58(8):2223-2236.
19 Xu Z,Ren T,Xiao C,et al.Nickel promotes the invasive potential of human lung cancer cells via TLR4/MyD88 signaling.Toxicology,2011,285(1-2):25-30.
20 Ikebe M,Kitaura Y,Nakamura M,et al.Lipopolysaccharide(LPS)increases the invasive ability of pancreatic cancer cells through the TLR4/MyD88 signaling pathway.J Surg Oncol,2009,100(8):725-731.
21 Yang L,Seki E.Toll-Like Receptors in Liver Fibrosis:Cellular Crosstalk and Mechanisms.Front Physiol,2012,3:138.
