慢病毒载体介导的RNA 干扰对HDAC2 基因表达的影响*
2015-04-28刘燕青黄韻祝娄方方孔维莹
刘燕青,黄 健,黄韻祝**,娄方方,孔维莹
(1.贵州医科大学 医学检验学院,贵州 贵阳 550004;2.贵州医科大学附院 生化科,贵州 贵阳 550004)
组蛋白去乙酰化酶2(HDAC2)属于I 类组蛋白去乙酰化酶,其催化结构含有与锌离子螯合的保守氨基酸残基,能调控细胞信号转导和基因表达[1]。HDAC2 在肿瘤组织中有异常表达,与肿瘤的发生、发展有关,在某些特定的肿瘤中HDAC2的删除可导致肿瘤细胞生长停滞和凋亡[2-5]。本课题组前期研究利用基因克隆技术构建了3 对针对HDAC2 基因的慢病毒重组表达质粒pLKO.1-HDAC2-shRNA 1、2、3,并经酶切鉴定和测序验证证实3 对重组质粒构建成功。本研究利用课题组构建的HDAC2 基因重组慢病毒表达质粒pLKO.1-HDAC2-shRNA,制备高浓度的慢病毒颗粒,感染人肝癌细胞株HepG2,观察HDAC2 基因的mRNA 和蛋白水平表达,从而判断其干扰效率,为后续研究HDAC2 基因的功能及作用机制奠定基础。
1 材料与方法
1.1 实验材料
pLKO.1-puro 慢病毒表达载体与psPAX2 及pMD2.G 质粒(美国addgene 公司),人肝癌HepG2细胞株与stbl2 感受态细胞(课题组保存),人胚肾293T 细胞(武汉大学典型保藏中心),20 bp DNA Ladder 与SYBR®Premix Ex TaqTMII(日本Takara公司),胎牛血清及DMEN 高糖培养基(Gibco 公司),阳离子脂质体LipofectamineTM2000、opti-MEM(Invitrogen 公司),鼠抗人HDAC2 单克隆抗体(Abcam 公司),LaminB1 抗体(武汉三鹰),细胞核与细胞浆蛋白提取试剂盒、辣根过氧化物酶标记的山羊抗兔及山羊抗小鼠IgG 二抗和BCA 蛋白浓度测定试剂盒(碧云天),PVDF 膜(Millipore 公司),荧光倒置显微镜(NIKON),恒温培养箱(日本三洋)。其他试剂均为国产分析纯。
1.2 方法
1.2.1 细胞培养 HepG2 细胞与293T 细胞复苏后常规培养于含100 mL/L 胎牛血清(FBS)的高糖DMEM 培养基,37 ℃、50 mL/L CO2饱和湿度条件下培养。取对数生长期的细胞进行实验。
1.2.2 筛选质粒与转染试剂最适比例 先对脂质体的转染条件进行优化,选用理论上不会影响细胞生长的对照质粒pEGFP-N1。将质粒pEGFP-N1 转化stbl2 感受态细胞,用含100 mg/L 卡那霉素的LB 培养基培养质粒pEGFP-N1,挑选单克隆鉴定,摇菌扩大、无内毒素抽提。取对数生长期293T 细胞铺6 孔板,1×105/孔,培养24 h 使细胞汇合度达70%~90%。实验分5 组,即质粒pEGFP-N1(μg)与脂质体Lipofectamine2000(μL)以1 ∶1、1∶2、1∶3、1∶4及1∶5的比例转染人胚肾细胞293T,分别取5 份3 μg pEGFP-N1 溶解至500 μL opti-MEM 中,分别吸取3 μL、6 μL、9 μL、12 μL、15 μL Lipofectamine2000 加入到500 μL opti-MEM,室温孵育5 min,然后将各组Lipofectamine2000 稀释液加到相应的质粒中,室温静置20 min,将混合物逐滴加入6 孔板,37 ℃、50 mL/L CO2培养8 h 后换成完全培养基,继续培养24 h、48 h,荧光显微镜下观察绿色荧光蛋白表达情况。
1.2.3 慢病毒包装 将前期构建的重组质粒pLKO.1-HDAC2-shRNA 与包装质粒psPAX2 和包膜质粒pMD2.G 采用脂质体介导法转染到293T 细胞中,包装成慢病毒颗粒。于100 mL/L 胎牛血清的DMEM 培养基内,置37 ℃、50 mL/LCO2培养箱中培养。去内毒素大提各组重组质粒pLKO.1-HDAC2-shRNA 和包装质粒psPAX2 和包膜质粒pMD2.G。转染前1 天,胰酶消化对数生长期293T细胞,用无抗生素培养基接种25 cm2培养瓶,8×105/瓶培养过夜,使细胞密度达50%~60%汇合度,进行转染。质粒(μg)∶脂质体(μL)按最优比例,将各组质粒(空载pLKO.1、重组质粒pLKO.1-HDAC2-shRNA1、2、3、neg 8 μg,psPAX2 6 μg,pMD2.G 2 μg)与低血清Opti-MEM 培养基在EP管中稀释至500 μL,取另一EP 管稀释48 μL 脂质体Lipofectamine2000 于Opti-MEM 培养基至500 μL,室温孵育5 min,将含Lipofectamine2000 的混合液加入质粒混合液中混匀,室温静置20 min。用无血清无抗生素DMEM 洗涤293T 细胞两遍,加入4 mL无血清无抗生素DMEM,然后将1 mL 质粒DNA-脂质体复合物逐滴加到细胞培养瓶中,37 ℃、50 mL/LCO2培养8 h 后,换成无抗生素含100 mL/L 胎牛血清的完全培养基继续培养;以pEGFP-N1 质粒同步转染检测转染效率。转染培养24 h 后更换为完全培养基,48 h 收集含慢病毒颗粒的细胞上清液,4 ℃保存。更换新培养基继续培养,24 h 后第2 次细胞上清液,与第1 次收集的上清液混合,室温2 000 r/min离心5 min 去除细胞碎片,收集病毒上清液,再经0.45 μm 滤器过滤病毒上清液,分装,-80 ℃保存备用。
1.2.4 感染靶细胞及细胞形态观察 取处于对数生长期肝癌HepG2 细胞,以不含抗生素100 mL/L胎牛血清加DMEM 接种至6 cm 培养皿,7×105个/皿,37 ℃,50 mL/LCO2培养24 h,细胞融合率为70%~90%时进行感染。实验分为6 组:未处理HepG2 细胞组(空白对照组)、空载体对照组(pLKO.1)、阴性对照组(pLKO.1-HDAC2-shRNAneg)、pLKO.1-HDAC2-shRNA 1 组、pLKO.1-HDAC2-shRNA 2 组、pLKO.1-HDAC2-shRNA 3 组。空白对照组不加入任何病毒颗粒换液后继续培养,空载体对照组、阴性对照组和pLKO.1-HDAC2-shRNA 1、2、3 组细胞中分别加入重组慢病毒颗粒进行细胞感染处理。各感染组先用无血清无抗生素DMEM 洗涤HepG2 细胞两遍,加入3 mL 无血清无抗生素DMEM,取适量polybrene 加入上述培养基中使其终浓度为5 mg/L,然后将各组2 mL 病毒液逐滴加到细胞培养皿中混匀;37 ℃,50 mL/LCO2培养24 h 后换成无抗生素含100 mL/L 胎牛血清的完全培养基继续培养;感染48 h 提取RNA 采用实时荧光定量PCR 分析感染后HDAC2 mRNA 水平,感染72 h 后提取细胞核蛋白进行Western blotting 分析。
1.2.5 HDAC2 mRNA 的检测 pLKO.1-HDAC2-shRNA 慢病毒感染HepG2 细胞48 h 后,提取各组细胞的总RNA,反转录为cDNA 为模板应用实时荧光定量PCR 检测HDAC2 mRNA 的表达情况。HDAC2 基因PCR 引物由上海捷瑞公司合成,上游引物为5'-ATTACTGATGCTTGGAGGAGGT-3',下游引物为 5'-TATTCTGGAGTGTTCTGGTTTG-3';以GAPDH 为内参,上游引物为 5'-CGCTGAGTACGTCGTGGAGTC-3',下游引物为5'-GCTGATGATCTTGAGGCTGTTGTC-3'。反应条件为95 ℃30 s,1 个循环;95 ℃5 s,60 ℃30 s,40 个循环;95 ℃,5 s,60 ℃1 min,95 ℃,1 个循环;50 ℃30 s,1 个循环。每组设3 个复孔,实验重复3 次。以HepG2 细胞组为空白组,根据荧光信号得到Ct 值,根据公式2-△△Ct计算HDAC2 mRNA 的相对表达量。
1.2.6 HDAC2 蛋白的检测 取感染后72 h 实验组和对照组细胞,用4 ℃预冷PBS 洗涤细胞,收集细胞并提取细胞核蛋白,BCA 法测定蛋白质浓度,分装,-80 ℃保存。取20 μg 蛋白加适量2×SDS上样缓冲液,混匀,沸水浴5 min 预变性,冰上冷却。经10%的SDS-PAGE 电泳分离后,采用湿转法电转移250 mA、2 h,将蛋白转移至PVDF 膜上,5%脱脂奶粉封闭1 h,加入HDAC2 一抗(1∶5 000)4 ℃缓慢振摇过夜,TBST 洗膜6 次,5 min/次,加入辣根过氧化物酶标记的二抗(1 ∶500),室温孵育1 h,洗膜后在暗室内用增强型化学发光(ECL)发光显色,胶片曝光。取胶片扫描获取图像,计算条带的灰度值作为蛋白的表达量。以LaminB1 作内参照,实验重复3 次。
1.3 统计学处理
采用SPSS 16.0 统计软件进行统计分析,计量资料用均数±标准差)表示,组间比较采用完全随机设计的单因素方差分析,检验水准α=0.05,P <0.05 为差异具有统计学意义。
2 结果
2.1 质粒与转染试剂比例优化
当质粒∶Lipofectamine2000 为1∶3时,转染效率能达70%以上。见图1。
2.2 重组慢病毒颗粒感染后HepG2 细胞形态变化
感染HepG2 细胞72 h 后,在倒置相差显微镜下见到对照组HepG2 细胞生长分裂活跃,形态规则,胞核椭圆且位于细胞中央。pLKO.1-HDAC2-shRNA 1、2、3 组HepG2 细胞皱缩,形态多样,细胞连接消失,胞质密度增加,细胞核固缩,染色质致密,并边集于核膜内侧,胞质中有空泡形成,有凋亡小体产生,可见细胞明显被抑制。见图2。
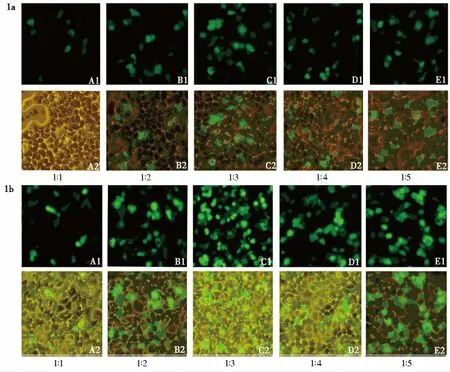
图1 pEGFP-N1 转染293T 细胞的荧光表达(200×)Fig.1 Fluorescent observation of EGFP expression of 293T cells after transfection with pEGFP-N1 vector
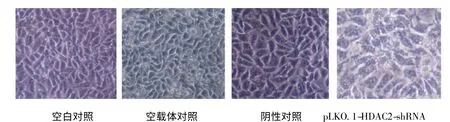
图2 自然光下感染的HepG2 细胞形态(72 h)Fig.2 Inverted microscope observation of cell morphology of HepG2 cells 72h in natural light after transfection with recombinant plasmid pLKO.1-HDAC2-shRNA
2.3 慢病毒颗粒感染HepG2 细胞后HDAC2 mRNA 表达及抑制率
慢病毒颗粒感染HepG2 细胞48 h 后,HDAC2基因表达在空载体对照组、阴性对照组与HepG2空白组间,差异无统计学意义(P >0.05);pLKO.1-HDAC2-shRNA 1 组、pLKO.1-HDAC2-shRNA 2 组、pLKO.1-HDAC2-shRNA 3 组与空白对照组、空载体对照及阴性对照组比较,HDAC2 基因mRNA 表达量均有下降,差异有统计学意义(P <0.05);其中pLKO.1-HDAC2-shRNA 1 组对HDAC2 mRNA 的抑制率最高,达到82.09%。见表1 和图3。
2.4 慢病毒颗粒感染HepG2 细胞72 h HDAC2 蛋白表达
HepG2 细胞经慢病毒感染72 h 后,在HepG2空白对照、空载体对照及阴性对照组均可见明显的HDAC2 蛋白表达,但3 组间差异无统计学意义(P>0.05);包装的含pLKO.1-HDAC2-shRNA 重组质粒的慢病毒颗粒感染细胞后,pLKO.1-HDAC2-shRNA 1、2、3 组HepG2 细胞中HDAC2 蛋白条带均较空白对照组、空载体对照组及阴性对照组明显减弱,组间比较差异有统计学意义(P <0.05),见图4、图5;pLKO.1-HDAC2-shRNA 1 组和3 组比pLKO.1-HDAC2-shRNA 2 组的条带更弱,对蛋白表达的干扰效果更好(P <0.05);HDAC2-shRNA 1组与HDAC2-shRNA 3 组比较,差异无统计学意义(P >0.05)。
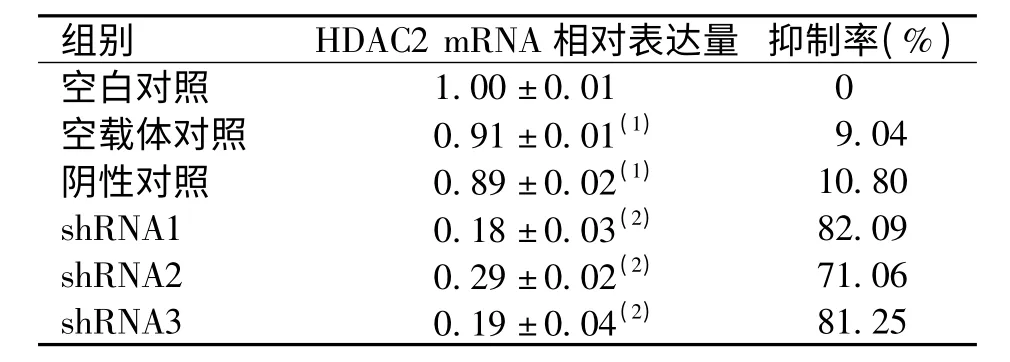
表1 shRNA 对HDAC2 mRNA 的抑制率Tab.1 The suppression rate of HDAC2 mRNA with shRNA

图3 各组HDAC2 mRNA 相对表达量Fig.3 Relative expression levels of HDAC2 mRNA in the six groups

图4 pLKO.1-HDAC2-shRNA 转染后HepG2 细胞中HDAC2 蛋白表达(Western blotting)Fig.4 Protein expression of HDAC2 detected by Western blotting analysis in HepG2 cells transfected with pLKO.1-HDAC2-shRNA
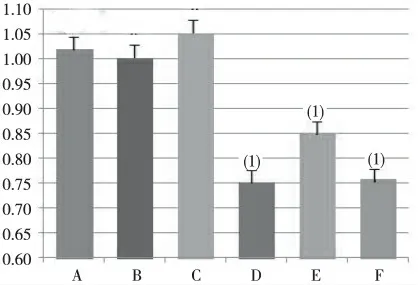
图5 HepG2 细胞HDAC2 蛋白的表达Fig.5 Protein expression of HDAC2 detected by gray value analysis in HepG2 cells transfected with pLKO.1-HDAC2-shRNA
3 讨论
从表观遗传学来讲,肿瘤的发生主要包括DNA 甲基化、组蛋白乙酰化、染色体重塑和非编码RNA 调控等。细胞及有机体内组蛋白的乙酰化和去乙酰化之间存在一种受到严格控制的动态平衡,是调节基因表达的一个重要因素,这种平衡的打破成为癌症产生的直接诱因[6]。HDACs 可被募集结合在特定的启动子区,从而导致许多基因转录受抑制,抑制抑癌基因的表达,因此HDACs 可能成为肿瘤治疗的靶点。HDAC2 处于信号转导途径的下游,有可能在染色体翻译修饰后对转录进行快速调节。研究发现,HDAC2 过表达与p21 的表达降低密切相关,抑制HDAC2 表达能上调p21 的表达和下调细胞周期蛋白cyclinD1 和cyclinA 的表达,提示HDAC2 在维持代谢平衡、调控细胞周期及肿瘤生成中可能发挥着重要作用[7-10]。研究发现,HDAC2 可通过多种机制达到促癌作用,而HDAC2的敲除可导致某些肿瘤细胞生长停滞和凋亡;另外,HDAC2 也可通过抑制抑瘤因子p53,p21 和促进原癌基因MYC 的表达,从而建立阻碍细胞凋亡和细胞周期停滞的恶性循环[11]。
RNA 干扰(RNAi)是在基因的转录后针对含有特异性序列的mRNA 进行沉默[12],RNAi 技术广泛应用于基因功能分析、信号转导通路研究和基因治疗。由于siRNA 不能在细胞内长期存在,无法实现长时间稳定的RNA 干扰,慢病毒载体则解决了此问题[13]。慢病毒属于逆转录病毒之一,是一种复制缺陷型病毒载体,以HIV21 为基础发展而得,在宿主细胞内能以自身RNA 为模板在自身反转录酶的作用下合成cDNA,再以碱基配对原则合成双链DNA,经环化后整合到宿主细胞的染色体上并长期表达。目前关于经慢病毒载体介导的目的基因沉默研究也日趋成熟。在本课题组前期研究中,通过利用基因克隆技术,成功构建了3 条pLKO.1-HDAC2-shRNA 干扰质粒载体和一条阴性对照载体。采用pLKO.1 慢病毒表达系统,可降低脂质体转染效率低,瞬时转染半衰期短的问题,且安全性高、操作性简单。在本实验中,由于质粒和脂质体浓度都会不同程度地影响细胞的生长和增殖,为了同时满足高效转染率和低细胞毒性,首先利用对细胞无毒性的pEGFP-N1 质粒,选取几个比例的质粒与脂质体Lipofectamine2000 转染慢病毒包装细胞293T,24 h、48 h 后荧光显微镜下观察细胞绿色荧光信号表达强度,获得最适的质粒与转染试剂比例进行下一步实验。本实验结果显示,当质粒:Lipofectamine2000 为1∶3时,转染效率最高,所以本研究的后续实验根据比例来进行瞬时转染靶细胞以及包装慢病毒能达到很好的效果。慢病毒颗粒转染哺乳动物细胞的效率比质粒载体高,本实验首先运用脂质体法介导的转染将构建成功的pLKO.1-HDAC2-shRNA 干扰质粒载体及阴性对照转染293T 细胞包装慢病毒颗粒,再感染HepG2 靶细胞,提取总RNA 和蛋白进行验证沉默效率。Real-time PCR 结果显示,HDAC2-shRNA 能有效的抑制HepG2 细胞中HDAC2 mRNA 的表达,其中HDAC2-shRNA1 对HDAC2 基因的抑制率高达82.09%,HDAC2-shRNA2 与 HDAC2-shRNA3 对HepG2 细胞 HDAC2 基因的抑制率分别是71.06%、81.25%,说明构建的HDAC2-shRNA 重组质粒在转录水平能有效的抑制HDAC2 基因的表达。Western blotting 结果显示,感染72 h 后,构建的HDAC2 shRNA 能有效地抑制HepG2 细胞中的HDAC2 蛋白表达,与空白对照、空载体对照及阴性shRNA 对照组比较,差异明显,而空白对照、空载体对照及阴性shRNA 对照组之间表达差异不显著,提示构建的HDAC2-shRNA 能有效下调肝癌HepG2 细胞中HDAC2 蛋白的表达,进一步证明成功构建HDAC2-shRNA 干扰载体,该研究为后续进行慢病毒包装及HDAC2 在肝癌中的功能提供理想的研究平台。
综上所述,本实验采用分子克隆技术成功构建了pLKO.1-HDAC2-shRNA 慢病毒载体,并证实了其在肝癌HepG2 细胞中对HDAC2 基因的干扰效果,提示其可有效地沉默肝癌HepG2 细胞中HDAC2 基因的表达,说明HDAC2-shRNA 基因靶点筛选构建的特异性和有效性,该研究为后续进行HDAC2 在肝癌中的功能提供了良好的前期实验基础。
[1]Wang SS,Li XM,Parra M,et al.Control of endothelial cell proliferation and migrationby VEGF signaling to histone deacetylase[J].Proc Natl Acad Sci USA,2008(22):7738-7743.
[2]Bracker TU,Sommer A,Fichtner I,et al.Efficacy of MS-275,a selective inhibitor of class I histone deacetylases,in human colon cancer models.Int J Oncol,2009(35):909-920.
[3]Weichert W.HDAC expression and clinical prognosis in human malignancies[J].Cancer Lett,2009(2):168-176.
[4]Adams H,Fritzsche FR,Dirnhofer S,et al.Class I histone deacetylases 1,2 and 3 are highly expressed in classical Hodgkin’s lymphoma[J].Expert Opin Ther Targets,2010(6):577-584.
[5]Fritsche P,Seidler B,Schuler S,et al.HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA[J].Gut,2009(10):1399-1409.
[6]Kouzarides T.Histone acetylases and deacetylases in cell proliferation[J].Curr Opin Genet Dev,1999(1):40-48.
[7]管晓翔,陈龙邦.组蛋白乙酰化修饰在基因表达调控中的作用机制[J].中华肿瘤防治杂志,2007(4):307-310.
[8]Yamaguchi T,Cubizolles F,Zhang Y,et al.Histone deacetyIases 1 and 2 act in concert to promote the G1-to-S progression[J].Genes Dev,2010(5):455-469.
[9]Huang BH,Laban M,Leung CH,et al..Inhibition of histone deacetylase 2 increases apoptosis and p21 CIP1/WAF1 expression.independent of histone deacetylase 1[J].Cell Death Differ,2015(4):395-404.
[10]Hrzenjak A,Moinfar F,Kremser ML,et al.Valproate inhibition of histone deacetylase 2 affects differentiation and decreases proliferation of endometrial stromal sarcoma cell[J].Mol Cancer Ther,2006(9):2203-2210.
[11]Witt O,Deubzer HE,Milde T,et al.HDAC family:What are the cancer relevant targets[J].Cancer Lett,2009(1):8-21.
[12]Elbashir SM,Harborth J,Lendeekel WY,et al.Duplexes of 21-nucleotide RNAs mediate RNA interferenee in cultured mammalian cells[J].Nature,2001(1):494-498.
[13]Hannon G J.RNA interference[J].Nature,2002(6894):244-251.
