超高效液相色谱法检测6 种探针底物代谢产物并评价人细胞色素P450 同工酶活性*
2015-04-28谢玉敏王永林
陆 苑,谢玉敏,潘 洁,黄 勇,王永林**
(1.贵州医科大学 贵州省药物制剂重点实验室,贵州 贵阳 550004;2.贵州医科大学 药学院 民族药与中药开发应用教育部工程研究中心,贵州贵阳 550004)
随着代谢性药物相互作用引起的不良反应案例逐年增多,对于药物间的相互作用的有效预测就显得尤为必要。研究表明,导致药物代谢性相互作用的主要原因是CYP450 酶系被抑制或被诱导,其中酶抑制作用所致药物相互作用的临床意义远大于酶诱导作用,约占代谢性药物相互作用的70%。酶抑制作用的研究最常使用的模型就是肝微粒体[1-4]。因此,本文建立选用甲苯磺丁脲(CYP2C9)、氯唑沙宗(CYP2E1)、咪达唑仑和睾酮(CYP3A4)、奥美拉唑(CYP2C19)和对乙酰氨基酚(CYP2A1)的作为探针底物,以其各自专一性代谢产物作为UPLC-MS/MS 检测对象来评价5 种CYP450 酶的活性,并通过方法学验证,为体外快速预测潜在的药物相互作用提供参考。
1 实验仪器与材料
超高效液相色谱系统(ACQUITY UPLC,美国沃特世公司,Allegra 64R 低温高速离心机(美国Beckman Coulter 公司),700 系列超低温冰箱(美国Thermo 公司)。非那西丁(批号81105)、奥美拉唑(批号90925)、甲苯磺丁脲(批号20321)、睾酮(批号10519)、磺胺苯吡唑(批号01022)和酮康唑(批号11205)均购于德国Dr 公司,咪达唑仑注射液(批号20120903)购于江苏恩华药业股份有限公司,氯唑沙宗(批号100364-200301)、对乙酰氨基酚(批号100018-200408)和氟康唑(批号100314-201204)购于中国食品药品检定研究院,羟基甲苯磺丁脲(批号1-PSB-27-2)、5 羟基奥美拉唑(批号1-PSB-27-2)、6-羟基氯唑沙宗(批号6-QFY-28-2)和6β-羟基睾酮(批号KIT0635)购于加拿大TRC 公司,α-羟基咪达唑仑(批号FN101512-07)购于美国Cerilliant 公司,氯美噻唑(批号4300-133)fluorochem,α-萘黄酮(批号AO222928)购于ACROS ORGANICS,人肝微粒体购于美国BD 公司。
2 方法和结果
2.1 体外代谢实验
采用NADPH 再生系统,终体积为200 μL 的体外孵育体系包括含肝微粒体蛋白0.5 g/L、NADP+20 g/L、六磷酸葡萄糖20 g/L、MgCl213.3 g/L 及六磷酸葡萄糖脱氢酶40 U/mL,甲苯磺丁脲、氯唑沙宗、咪达唑仑、睾酮、奥美拉唑和非那西丁在溶液中的浓度分别为100、100、50、200、50 和50 μmol/L,其余为pH7.4 的0.1 mol/L PBS 缓冲溶液。37 ℃预孵3 min,加入底物开始反应。每次加入反应体系中有机试剂终浓度不超过1% (v/v)。反应完毕后加入100 μL 甲醇终止反应,再加入2 mg/L 葛根素溶液100 μL,涡混,超声3 min,15 000 r/min离心10 min,取上清液1 μL 供UPLC-MS/MS 分析。
2.2 分析条件
2.2.1 液相条件 色谱柱Waters BEH C18(2.1 mm×50 mm,1.7 μm)柱,流速0.35 mL/min,柱温45 ℃,流动相0.1%甲酸乙腈(A)-0.1%甲酸水溶液(B),检测6 种专一性代谢产物的梯度条件0 ~3.0 min 5%~65%A,3.0 ~3.5 min 65%~90%A,3.5~4.5 min 10%A;进样体积1 μL。
2.2.2 质谱条件 电喷雾电离源(ESI);毛细管电压3 kV,离子源温度120 ℃去溶剂气温度350 ℃;去溶剂气N2,流速650 L/h;反吹气N2,流速50 L/h;碰撞气Ar,流速0.16 mL/min;质谱数据采集及处理软件为MassLynx V4.1 工作站,扫描方式为多反应离子监测模式(MRM)。离子对质谱条件见表1。
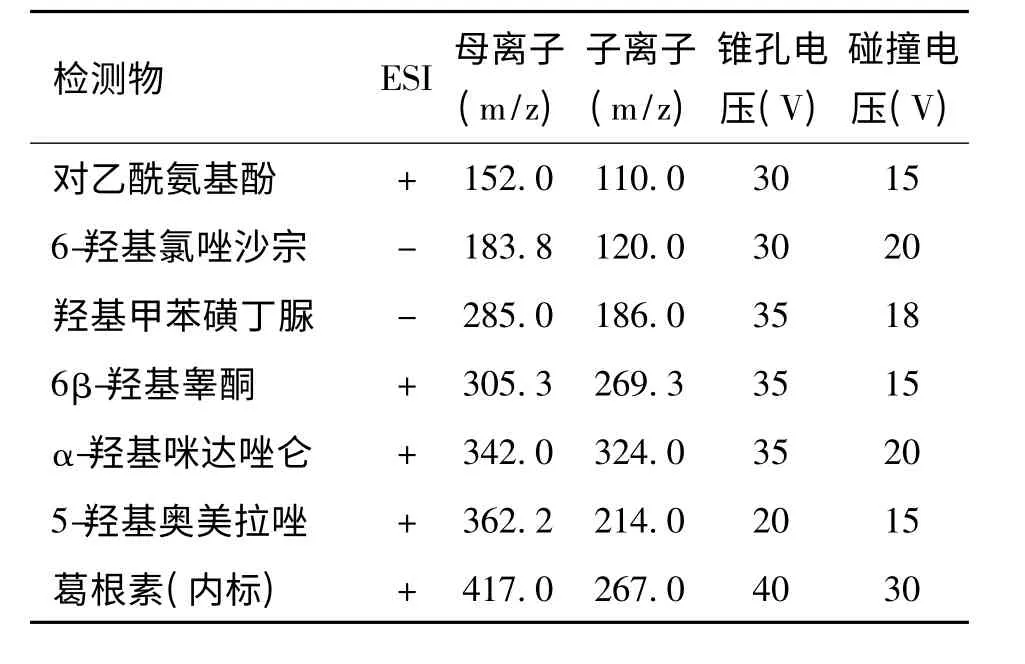
表1 离子对质谱条件Tab.1 Ion pair mass spectrometry
2.3 方法学考察
2.3.1 专属性 分别取6 份人肝微粒体孵育液,除不加探针药物和内标外,其余按“2.1”项操作,获得空白样品色谱图1-A;将一定浓度标准溶液和内标溶液加入灭活的人肝微粒体孵育液(置于沸水浴中10 min),同法得色谱图1-B;各探针药物在人肝微粒体孵育液中孵育60 min,同法得色谱图1-C。结果见图1,对乙酰氨基酚、内标葛根素、6-羟基氯唑沙宗、5-羟基奥美拉唑、羟基甲苯磺丁脲、6β-羟基睾酮和α-羟基咪达唑仑的相对保留时间(RT)分别为0.75、1.00、1.23、1.34、1.79、1.84和1.91 min。结果表明,空白肝微粒体无干扰,专属性良好。

图1 人肝微粒体孵育液体系色谱图Fig.1 Chromatographic chart of incubation fluid system of human liver microsomes
2.3.2 标准曲线的制备和最低检测限 将人肝微粒体灭活后,按“2.1”项下操作,加入含有6 种代谢产物的混合标准溶液。以待测物的峰面积与内标峰面积之比(A/Ai)为纵坐标Y,各物质浓度(C)为横坐标X 用加权最小二乘法进行线性回归,权重系数为1/X,求得标准曲线。6 种探针药物的代谢产物的最低检测限(LLOD)定义为S/N≥3,各成分相关系数(R2)>0.996。见表2,在相应范围内,6 种代谢物的线性关系良好。
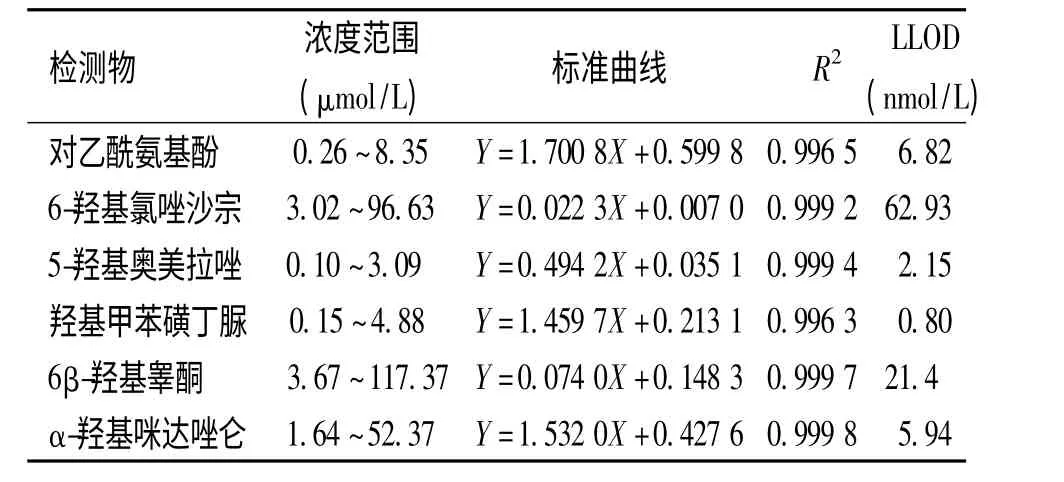
表2 6 种代谢产物回归曲线方程Tab.2 Regression curve equation for 6 metabolites
2.3.3 准确度和精密度 按“2.3.2”项下分别配制低、中、高3 个浓度的质控(QC)样品,每个浓度平行6 份(n=6),连续测定3 d,根据当日标准曲线计算QC 样品的浓度,评价方法的准确度与日内、日间精密度。见表3。该方法准确、可靠、重现性好。
表3 检测物的的准确度、日内和日间精密度()Tab.3 Accuracy and precision of the detection
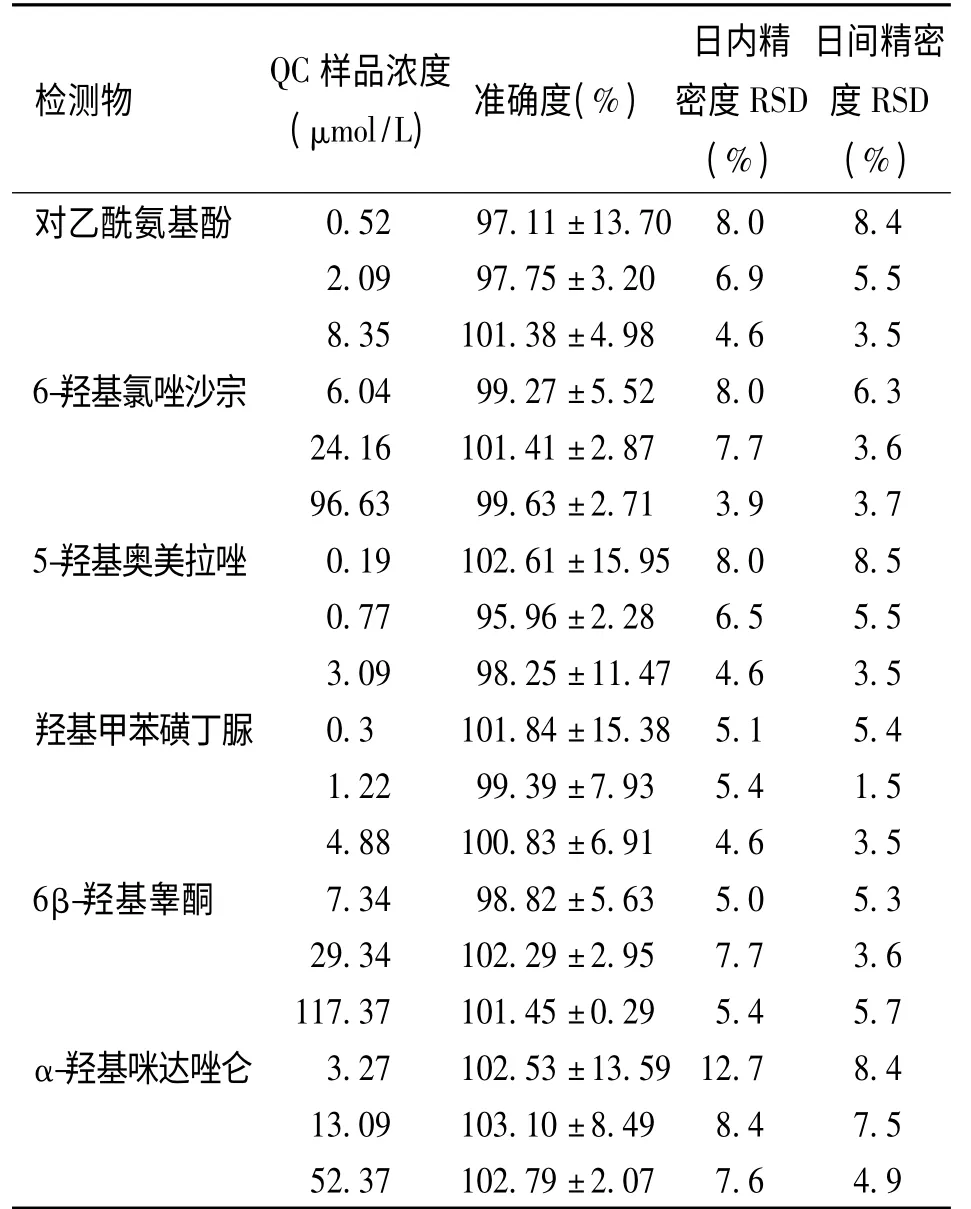
表3 检测物的的准确度、日内和日间精密度()Tab.3 Accuracy and precision of the detection
检测物Q(C μ 样m品ol/浓L)度 准确度(%)密日(度内%R精)S D日度(间%R精S)D密对乙酰氨基酚0.52 97.11±13.70 8.0 8.4 2.09 97.75±3.20 6.9 5.5 8.35 101.38±4.98 4.6 3.5 6-羟基氯唑沙宗 6.04 99.27±5.52 8.0 6.3 24.16 101.41±2.87 7.7 3.6 96.63 99.63±2.71 3.9 3.7 5-羟基奥美拉唑 0.19 102.61±15.95 8.0 8.5 0.77 95.96±2.28 6.5 5.5 3.09 98.25±11.47 4.6 3.5羟基甲苯磺丁脲 0.3 101.84±15.38 5.1 5.4 1.22 99.39±7.93 5.4 1.5 4.88 100.83±6.91 4.6 3.5 6β-羟基睾酮 7.34 98.82±5.63 5.0 5.3 29.34 102.29±2.95 7.7 3.6 117.37 101.45±0.29 5.4 5.7 α-羟基咪达唑仑 3.27 102.53±13.59 12.7 8.4 13.09 103.10±8.49 8.4 7.5 52.37 102.79±2.07 7.6 4.9
2.3.4 提取回收率和基质效应 按“2.3.2”项下操作,分别配制低、中、高3 个浓度的QC 样品,每个浓度平行操作5 份,按“2.1”项下操作(A 样品)。另取空白人肝微粒体孵育体系,除不加混合标准溶液与内标外,其余按“2.1”项下操作,向获得的上清液中加入相应低、中、高浓度的混合标准溶液和内标(B 样品);另取上述低、中、高浓度的混合标准溶液与内标,以初始流动相溶解(C 样品)。内标以同样方法进行考察。提取回收率计算方法为B 样品与A 样品色谱峰面积之比,基质效应计算方法为B 样品与C 样品的色谱峰面积之比。6 种代谢物的提取回收率和基质效应结果结果见表4,内标的提取回收率和基质效应分别为98.6%和94.8%。实验结果表明各指标成分提取回收率良好,无明显的基质效应。
表4 检测物的提取回收率和基质效应(,n=6)Tab.4 Extraction recovery and matrix effect of the detection
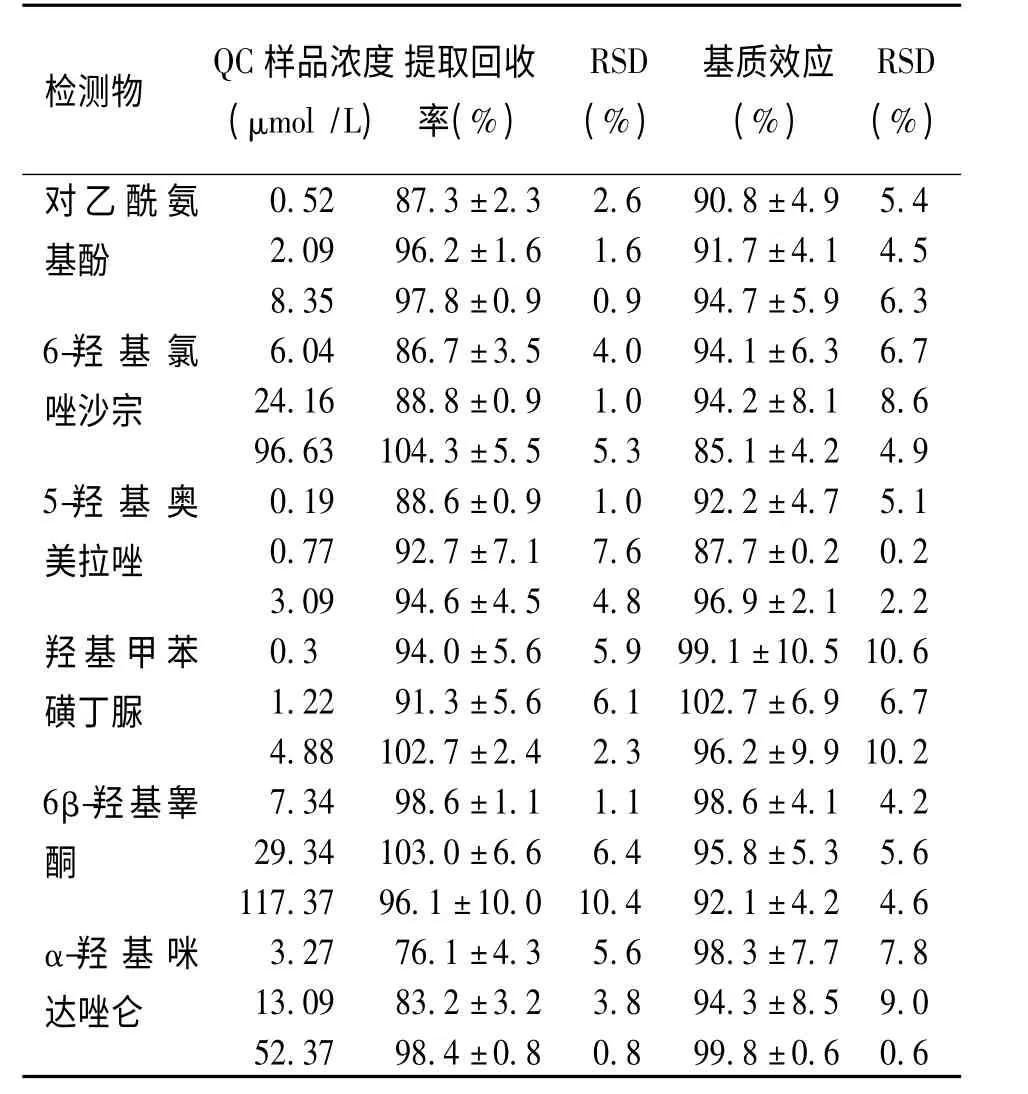
表4 检测物的提取回收率和基质效应(,n=6)Tab.4 Extraction recovery and matrix effect of the detection
检测物Q(C μ 样m o品l浓/L)度提率取(回%收)(R%S D)基(质%效)应(R%S D)对乙酰氨0.52 87.3±2.3 2.6 90.8±4.9 5.4基酚2 8..0 3 9 5 9 9 6 7..2 8±±1 0..6 9 1 0..6 9 9 9 1 4..7 7±±4 5..1 9 4 6..5 3 6-羟 基 氯6.04 86.7±3.5 4.0 94.1±6.3 6.7唑沙宗2 9 4 6..1 6 6 3 1 8 0 8 4..8 3±±0 5..9 5 1 5..0 3 9 8 4 5..2 1±±8 4..1 2 8 4..6 9 5-羟 基 奥0.19 88.6±0.9 1.0 92.2±4.7 5.1美拉唑0 3..7 0 7 9 9 9 2 4..7 6±±7 4..1 5 7 4..6 8 8 9 7 6..7 9±±0 2..2 1 0 2..2 2羟基甲苯0.3 94.0±5.6 5.9 99.1±10.5 10.6磺丁脲1 4..2 8 2 8 1 9 0 1 2..3 7±±5 2..6 4 6 2..1 3 10 9 2 6..7 2±±6 9..9 9 1 6 0..7 2 6β-羟基睾7.34 98.6±1.1 1.1 98.6±4.1 4.2酮1 2 1 9 7..3 3 4 7 1 9 0 6 3..1 0±±1 6 0..6 0 1 6 0..4 4 9 9 5 2..8 1±±5 4..3 2 5 4..6 6 α-羟 基 咪3.27 76.1±4.3 5.6 98.3±7.7 7.8达唑仑1 5 3 2..0 3 9 7 8 9 3 8..2 4±±3 0..2 8 3 0..8 8 9 9 4 9..3 8±±8 0..5 6 9 0..0 6
2.3.5 稳定性考察 按“2.3.2”项下分别配制低、中、高3 个浓度的QC 样品,每一浓度5 个样本分析,置于自动进样器中0、6 h,室温(约20 ℃)下放置6 h,4 ℃下冷藏8 h,冻融循环3 次。人肝微粒体孵育样品中6 种专一性代谢产物的在上述放置条件下稳定。
2.3.6 质量控制实验 在每批测定时随行测定低、中、高3 个浓度的QC 样品,每个浓度的QC 样品进行双样本分析,测定数不少于样本总量的5%。结果3 个批次实验的QC 样品中、高浓度的回收率为85%~115%,低浓度的回收率为80%~120%,符合生物样品分析方法的检测要求。
2.4 阳性抑制剂对CYP450 酶活性的影响
参照FDA 指南,选择5 种特异性抑制剂磺胺苯吡唑(CYP2C9)、氯美噻唑(CYP2E1)、酮康唑(CYP3A4)、氟 康 唑(CYP2C19)、α-萘 黄 酮(CYP2A1),分别取0、0.01、0.05、0.1、0.3、1.0、3.0 以及10.0 μmol/L8 种不同浓度加入到Cocktail探针底物反应体系中。按“2.1”项下操作,孵育和进样分析,测定生成的专一性代谢产物。采用GraphPad 5.0 软件进行非线性拟合,各抑制剂的IC50值在文献报导值的3 倍以内[3],在一定浓度范围内,随着阳性抑制剂浓度的增加,酶活性逐渐降低。根据文献[4-12],可知磺胺苯吡唑对CYP2C9 的IC50值为0.2 ~1.52 μmol/L,氯美噻唑对CYP2E1 的IC50值为12 μmol/L,酮康唑对CYP3A4(咪达唑仑)的IC50值为0.008 ~1.8 μmol/L,酮康唑对CYP3A4(睾酮)的IC50值为2 ~23.8 μmol/L,α-萘黄酮对CYP1A2 的IC50值为0.04 ~0.2 μmol/L。见表5 和图2。
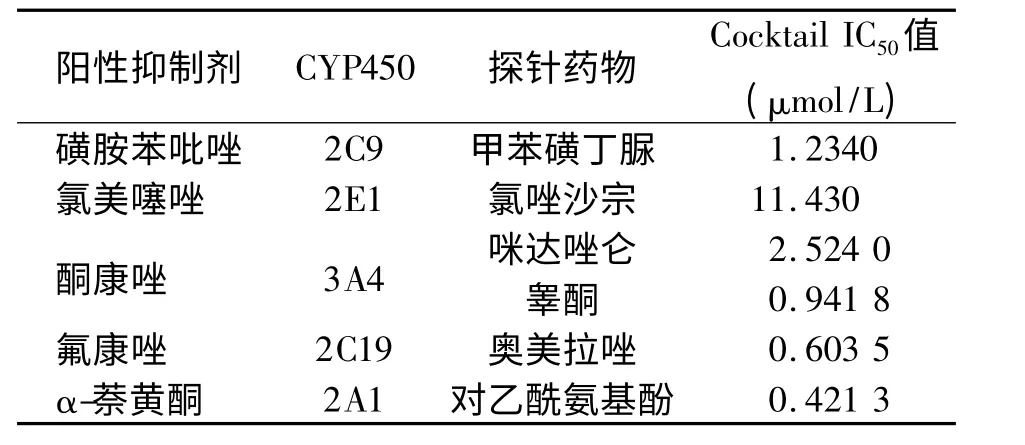
表5 特异性抑制剂的半数抑制浓度Tab.5 Median inhibitory concentrations of specific inhibitors
3 讨论
采用人混合肝微粒体作为研究对象,可降低个体和种属差异造成的影响,具有实际指导意义。本研究采用UPLC-MS/MS 仪器建立Cocktail 混合探针,可对药物的6 种代谢物起到灵敏、特异、可靠、超快速定量分析的方法,显著提高了定量分析的重复性、可靠性、准确性[13]。采用甲醇沉淀肝微粒体蛋白,高速离心净化样品,较常规的固相萃取法和有机溶剂萃取法可更经济、快速地满足高通量筛选的要求,以满足体外快速筛选药物相互作用。由于CYP3A4 具有底物依赖性,一般认为CYP3A4 潜在抑制能力评价的谨慎方法是使用多种探针来测定(至少两种探针底物),因此本研究选用了最常使用的咪达唑仑和睾酮[4]。
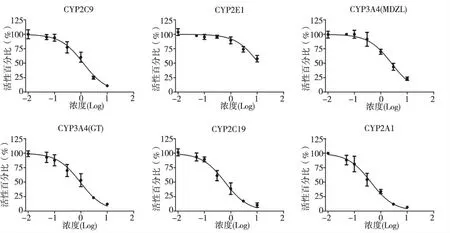
图2 特异性抑制剂对人肝微粒体细胞色素P450 酶5 种亚型的抑制曲线Fig.2 Inhibition curves of 5 subtypes of cytochrome P450 in human liver microsomes with specific inhibitors
因此,本方法既然可作为新药候选化合物的筛选工具,又可评价已上市药物及预测临床联合用药产生的药物相互作用;另外,还可评价中药提取物、单方或复方中成药、中药配伍对CYP450 的影响[14],预测潜在的药物相互作用,降低患者的用药风险,保障临床安全用药。
[1]Food and Drug Administration.Guidance for Industry Drug interaction studies-study design,data Analysis,and recommendations for dosing and labeling Recommendations[S].Silver Spring,MD 20993-0002:FDA,2012.
[2]叶林虎,孔令提,肖冰心,等.LC-MS/MS 同时测定5种探针底物代谢产物和快速评价细胞色素P450 同工酶的活性[J].中国药物警戒,2013(5):263-268.
[3]艾常虹,孙汉雄,李桦,等.中药有效成分对细胞色素P450 酶的抑制活性评价[J].中国药理学通报,2011(4):519-523.
[4]曾苏.药物代谢学[M].浙江:浙江大学出版社,2007.
[5]Turpeinen M,Jouko U,Jorma J,et al.Multiple P450 substrates in a single run:rapid and comprehensive in vitro interaction assay[J].European Journal of Pharmaceutical Sciences,2005(1):123-132.
[6]Lewis DF,Dickins M.Substrate SARs in human P450s[J].Drug discovery today,2002(17):918-925.
[7]Weaver R,Graham KS,Beattie IG,et al.Cytochrome P450 inhibition using recombinant proteins and mass spectrometry/multiple reaction monitoring technology in a cassette incubation[J].Drug Metabolism and Disposition,2003(7):955-966.
[8]Bu HZ,Knuth K,Magis L,et al.High-throughput cytochrome P450 inhibition screening via cassette probe-dosing strategy.IV.Validation of a direct injection on-line guard cartridge extraction/tandem mass spectrometry method for simultaneous CYP3A4,2D6 and 2E1 inhibition assessment[J].Rapid Commun Mass Spectrom,2000(20):1943-1948.
[9]Shader RI,Granda BW,Moltke LL,et al.Inhibition of human cytochrome P450 isoforms in vitro by zafirlukast[J].Bipharm Drug Dispos,1999(8):385-388.
[10]Rae Y,Soraya M,Xiao XW,et al.Evaluation of cytochrome P450 probe substrates commonly used by the pharmaceutical industry to study in vitro drug interactions[J].Drug Metabolism And Disposition,2002(12):1311-1319.
[11]Palmer JL,Scott RJ,Gibson A,et al.An interaction between the cytochrome P450 probe substrates chlorzoxazone(CYP2E1)and midazolam(CYP3A)[J].Br J Clin Pharmacol,2001(5):555-561.
[12]Kenworthy KE,Bloomer JC,Clarke SE,et al.CYP3A4 drug interactions:correlation of 10 in vitro probes substrates[J].Br J of Clin Pharmacol,1999(5):716-727.
[13]赵莹,冯怡,赵瑞芝.小鼠组织中龙胆苦苷含量的液相色谱一串联质谱测定[J].时珍国医国药,2010(7):1580-1582.
[14]窦志英,郑蓓蓓.Cocktail 探针药物法在中药研究中的应用[J].中国中医药信息杂志,2013(20):110-112.
