氮掺杂有序介孔碳-Ni纳米复合材料的制备及电化学性能
2015-04-01潘旭晨汤静薛海荣郭虎范晓莉朱泽涛何建平
潘旭晨 汤静 薛海荣 郭虎 范晓莉 朱泽涛 何建平
(南京航空航天大学材料科学与技术学院,南京210016)
氮掺杂有序介孔碳-Ni纳米复合材料的制备及电化学性能
潘旭晨 汤静 薛海荣 郭虎 范晓莉 朱泽涛 何建平*
(南京航空航天大学材料科学与技术学院,南京210016)
以F127为模板剂,NiCl2为镍源,尿素为氮源,间苯二酚甲醛原位聚合树脂为碳源,分别采用均相法和两相法制备Ni-NOMC-1,Ni-N-OMC-2纳米复合材料。X射线衍射(XRD)、激光拉曼以及透射电子显微镜(TEM)等测试结果表明,复合材料具有有序介孔结构,Ni以金属微粒形式嵌于碳骨架中,提高了有序介孔碳的石墨化程度。X射线光电子能谱测试(XPS)表明尿素热解后以4种形式存在:sp3杂化与C结合的N原子,吡啶N原子,sp2杂化与C结合的N原子以及quaternary-N原子。Ni-N的共改性改变了碳载体的理化性质,有利于Pt纳米粒子的负载与分散。均相法制备的Ni-N-OMC-1复合材料微波负载Pt后,氧还原极限电流密度为5.32 mA·cm-2,氢氧化电化学活性面积高达138.53 m2·g-1,电化学催化活性优于商业20%Pt/C材料(4.49 mA·cm-2, 96.98 m2·g-1)。
均相法;两相法;有序介孔碳;N-Ni复合掺杂;电催化活性
质子交换膜燃料电池(DMFC)依赖于高效的氧化与还原反应,将化学能转变为电能,其转化效率与电催化活性物质,电极载体等有关。在众多载体材料中,有序介孔碳由于具有丰富的孔道结构,较高的比表面积,且易于进行表面功能化修饰等优点而受到了广泛关注。目前较为成熟的制备有序介孔的方法有硬模板法和软模板法。相对而言,软模板法具有低价、高效且工业化生产更为便捷等优势。然而,采用软模板法制备的有序介孔碳,经惰性气体保护的高温热解后,碳表面活性基团严重缺失,有序介孔碳表面亲水性下降,不利于催化活性物质的有效负载与分散[1]。此外,由软模板法制备的有序介孔碳通常具有无定形碳孔壁,导电性能差,限制其在催化领域更广泛的应用。
通常采用非金属异质原子以及过渡金属对有序介孔碳进行改性。非金属异质原子(如N,P,S等)[2-4]的掺杂有助于提高碳材料的表面活性,其中氮是一种常用的掺杂元素,氮的引入改变了碳载体的理化性质,在碳载体表面形成的碱性位点与负载金属之间存在化学作用(如电子转移反应或配位反应)[5],从而有效抑制了金属微粒的团聚,促进了金属粒子在载体表面的均匀负载与分散。Datta等[6]以SBA-15为模板,乙二胺为氮源制备出高度晶化、导电能力较好的氮掺杂石墨壁有序介孔碳,金属Pt颗粒分散均匀,粒径不足5 nm,ICP测试结果表明Pt的担载量与载体中N含量有关。过渡金属的引入(如Fe,Co,Ni等)[7-9]有助于在较低温度(<1 000℃)下促使无定形碳转变为石墨化微晶。金属Ni是一种有效的催化石墨化金属,Tang等[10]在以软模板法制备有序介孔碳的过程中,将Ni盐添加至介孔碳前驱液中,700℃碳热还原后金属Ni微粒分散于介孔骨架中,有效催化了碳材料的石墨化转变,增强了碳载铂活性。Ni添加量为5%时,复合材料氢氧化的电化学活性面积是未添加金属时的13.9倍。
将氮源与过渡金属盐复合添加于介孔碳前驱体中,可能更有利于改善有序介孔碳的性能。我们课题组采用软模板法,以预聚合的酚醛树脂(Mr<500)为碳源,原位添加钴源和氮源,制备得到Co-NOMC复合材料,该试样的催化性能优于Co,N单独改性的Co-OMC以及N-OMC,但材料的介孔结构受到了一定破坏[11]。基于苯酚的酚醛树脂为线性聚合物,交联度低,因而与模板剂自组装形成的聚合物在碳化过程中容易坍塌。与苯酚相比,间苯二酚与甲醛分子的聚合活性更大,聚合物交联度更高,所得碳骨架的刚性得到加强。Wang[12]等以间苯二酚甲醛树脂为碳源,F127为模板剂,制备得到具有良好热稳定性的有序介孔碳。1 800℃时,介孔碳有序结构保持完好,孔道无明显收缩现象。为获得有序性较好的改性有序介孔碳材料,本文以间苯二酚甲醛原位聚合树脂为碳源,三嵌段共聚物F127为模板剂,NiCl2为镍源,尿素为氮源,分别采用均相法和两相法制备氮掺杂有序介孔碳-Ni纳米复合材料,并探究了微波负载Pt后材料的电化学性能。
1 实验部分
1.1 Ni-N-OMC-1,Ni-N-OMC-2复合材料的制备
(1)有序介孔碳(OMC)的制备方法如文献所述[13]。
(2)均相法:将1.0 g间苯二酚和0.03 g氢氧化钠溶解在16 g乙醇中,滴加2.2 g质量分数为37%的甲醛溶液,搅拌形成溶液A。将2 g F127,0.242 g氯化镍和0.10 g尿素溶解于12 g乙醇中,搅拌形成溶液B。将溶液A缓慢滴加至溶液B中,继续搅拌2 h,将此混合液转移至蒸发皿中室温下溶剂挥发24 h,然后将蒸发皿放于100℃真空干燥箱中热聚合24 h。最后将蒸发皿内形成的红褐色薄膜刮下,置于氮气氛围保护的管式炉中700℃碳化2 h得到目标产物,命名为Ni-N-OMC-1。
(3)两相法:将2 g F127溶解于20 g醇水混合溶液中(乙醇与水的体积比为1∶1),加入1.0 g间苯二酚,当间苯二酚完全溶解形成淡棕色溶液时,分别加入0.242 g的氯化镍以及0.10 g尿素,搅拌均匀后加入0.2 g质量分数为37%的盐酸为催化剂,搅拌2 h后,在上述混合液中滴加2.2 g甲醛溶液(质量分数37%),继续搅拌1 h至溶液变成浑浊状并逐渐分层,将分相后的产物静置4d后,弃去上清液,搅拌下层聚合产物至粘稠状,将其在85℃下聚合48 h。最后,将聚合产物置于氮气氛围保护的管式炉中700℃碳化2 h得到目标产物,命名为Ni-NOMC-2。
1.2 微波法制备Ni-N-OMC-1,Ni-N-OMC-2复合材料负载Pt催化剂
催化剂制备过程如下:将1.4 mL氯铂酸的乙二醇溶液(0.038 mol·L-1)与20 mL乙二醇混合均匀,用氢氧化钠的乙二醇溶液(2.5 mol·L-1)调节其pH值为9,将40 mg待负载材料(OMC,Ni-N-OMC-1,Ni-N-OMC-2)加入至上述Pt前驱体溶液中,超声波振荡30 min后将所得悬浮液置于微波炉中,调节功率至700 W,加热1 min,暂停10 s后再加热1 min。待混合物自然冷却后,用0.2 mol·L-1盐酸水溶液调节pH≈3,以促进Pt纳米粒子沉降。离心分离得到黑色产物,用乙醇和蒸馏水多次洗涤,80℃下真空干燥12 h即得目标产物。
1.3 材料的结构测试
采用德国BRUKER公司D8 ADVANCE X射线衍射仪进行XRD测试,阴极用Cu靶Kα线辐射(λ=0.154 178 nm),靶电流为50 mA,靶电压为30 kV,小角扫描范围为0.6°~10°,广角扫描范围为10°~90°,两种扫描方式的扫描速度均为2°·min-1;采用美国Micromeritics公司ASAP 2010型自动吸附仪,在液氮(77 K)条件下测定试样的N2吸脱附等温曲线及孔径分布分布曲线,试样的比表面积SBET按BET(Brunauer-Emmett-Teller)方程计算,孔径分布(D)按BJH(Barrett-Joyner-Halenda)模型计算;TEM测试采用FEI TecnaiG2型透射电子显微镜,加速电压为200 kV;激光拉曼测试采用法国JY HR800激光拉曼光谱仪;X射线光电子能谱测试(XPS)测试在PHI 5000 VersaProbe电子能谱仪上进行。
1.4 材料的电化学性能测试
电化学测试在工作站CHI660C上进行,电解液为0.5 mol·L-1H2SO4,测试采用三电极体系:涂覆催化剂的玻碳电极为工作电极,Pt电极为对电极,饱和甘汞电极为参比电极。催化剂工作电极的制备方法如下:将5.0 mg电催化剂置于5 mL离心管中,加入50 μL Nafion溶液,待其完全浸润催化剂后加入1.0 mL无水乙醇,超声波振荡30 min得到黑色悬浊液。用微量进样器移取25 μL黑色悬浊液缓慢滴于抛光的玻碳电极上,完全干燥后作为电催化性能测试的工作电极。循环伏安测试的电位区间为-0.22~0.98 V,扫速为20 mV·s-1。旋转圆盘电极(RDE)测试前电解液需用氧气饱和,转速为1 600 r· min-1,扫描速度为5 mV·s-1。
2 结果与讨论
2.1 试样的结构分析
如图1(a)所示,在2θ=0.7°~1.0°范围内的衍射峰对应于二维六方介孔结构(100)晶面。相比于OMC,试样Ni-N-OMC-1,Ni-N-OMC-2衍射峰向左偏移,这是由于金属镍盐在碳热过程中被还原为金属微粒,能够抑制碳化过程中孔道的收缩,因而试样Ni-NOMC-1,Ni-N-OMC-2具有较大的孔径,表现为衍射峰的左移。氯化镍与尿素的加入可能影响F127与树脂的自组装过程,一定程度上破坏介孔材料结构的有序性,但由于间苯二酚具有较强的反应活性,碳骨架刚性得到提高,因而试样Ni-N-OMC-1,Ni-NOMC-2仍具有明显的衍射峰。如图1(b)所示,试样Ni-N-OMC-1和Ni-N-OMC-2在44.3°,51.7°和76.1°处出现尖而窄的衍射峰,查阅衍射卡片可知它们分别对应于面心立方结构的金属Ni单质(111),(200)和(220)晶面[14]。样品Ni-N-OMC-1的前驱体溶液为碱性醇溶液体系,有利于Ni2+与间苯二酚-甲醛-尿素共缩聚树脂的羟基之间形成配合物,增强了Ni2+与聚合物之间的结合力,因而金属Ni流失量较小,结晶度较高,其大角衍射峰强度较大。两相法制备的Ni-N-OMC-2前驱体溶液为酸性的醇水混合体系,酸性体系下,Ni2+与间苯二酚-甲醛-尿素共缩聚树脂配合作用较弱,实验过程中发现相分离后的上层水清液呈现淡绿色,说明有一定量Ni2+溶解其中,从而使下层聚合物经干燥、碳热还原后所得的Ni-N-OMC-2复合材料中Ni的含量较低,对应的衍射峰强度较弱。根据Ni(111)晶面,由Scherrer方程计算得到Ni-N-OMC-1,Ni-N-OMC-2的平均粒径分别为38.5 nm和20.3 nm。试样的微观结构和颗粒分布情况可在TEM图中做进一步的观察和分析。
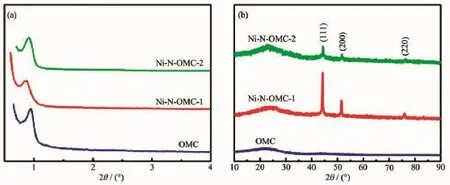
图1 样品的小角(a)和大角(b)XRD图Fig.1Small-angle XRD(a)and large-angle XRD(b)patterns of the samples
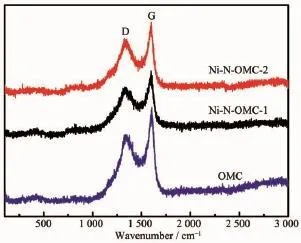
图2 样品的激光拉曼图谱Fig.2Raman spectra of the samples
激光拉曼光谱可以表征材料的石墨化程度。图2是样品OMC,Ni-N-OMC-1,Ni-N-OMC-2的拉曼图谱。图中在1 360 cm-1和1 580 cm-1左右出现的2个峰分别对应于无定形碳的无序峰(D峰)和石墨化碳的sp2杂化振动峰(G峰)。两峰的积分面积比值R (R=ID/IG)越小说明石墨化程度越高[15]。各样品的R值列于表1中。样品Ni-N-OMC-1中Ni负载量大,结晶度高(图1b),催化石墨化能力更强,因而Ni-NOMC-1(R:1.38)石墨化程度大于Ni-N-OMC-2(R:1.50),两者石墨化程度均大于未引入Ni的OMC(R:1.62)。相比于无定形碳,石墨化碳具有较高的热稳定性以及优异的抗氧化能力。此外,石墨化碳的π电子离域体系与Pt的5d空轨道之间具有电子效应,能够在一定程度上提高Pt催化剂的稳定性[16-17]。
图3为OMC,Ni-N-OMC-1,Ni-N-OMC-2纳米复合材料的N2吸附/脱附等温线以及由脱附曲线得到的孔径分布图。吸脱附等温线呈现LangmuirⅣ型,在相对分压p/p0为0.4~0.7之间具有1个明显的滞后环,这是由毛细管效应引起氮气在介孔中凝结所致,表明合成的碳材料具有均一的介孔孔道结构[18]。样品的孔结构参数列于表1中,与小角XRD衍射峰强弱顺序一致,样品比表面积大小顺序为OMC (602 m2·g-1)>Ni-N-OMC-2(592 m2·g-1)>Ni-N-OMC-1 (567 m2·g-1)。规则的孔结构以及高比表面积能够提供更多的活性点及存储电解液的空间,有利于提高传质速度,增大三相反应界面。孔径大小顺序为:Ni-N-OMC-1(3.9 nm)>Ni-N-OMC-2(3.5 nm)>OMC(3.1 nm),推断原因为金属Ni纳米粒子的嵌入增强了碳骨架的机械强度,有效抑制了孔道收缩现象。而金属Ni在催化无定形碳壁向类石墨化态转变的过程中,促使碳层排列得更加紧密,是孔径变大的又一原因[19]。作为电催化剂Pt载体,孔径的增大有利于电解液的渗透和离子的传输,减小了传质阻力。

表1 样品的孔结构参数Table 1Structure parameter of the samples
TEM测试进一步表征了OMC,Ni-N-OMC-1,Ni-N-OMC-2复合材料的微观结构。如图4(a)所示,OMC具有清晰的有序介孔孔道,由高倍透射图(b)可知,碳壁主要由无定形碳组成。如图4(c)所示,试样Ni-NOMC-1中,金属Ni纳米粒子均匀分布于介孔网格中。在图4(d)中可见环绕在Ni颗粒周围清晰的碳层,碳层间距为0.334 nm,为标准的石墨化碳层,进一步证明了在热处理阶段,金属Ni能有效催化碳由无定形态向类石墨化态转变。催化生成的石墨碳层提高了金属颗粒的化学稳定性,能够有效抑制团聚反应的发生,因而有助于获得粒径较小,分散均匀的Ni纳米粒子。如图4(c)所示,试样Ni-N-OMC-2中Ni微粒负载量低,平均粒径较小,由图2拉曼光谱可知该样品中金属Ni的催化石墨化反应能力较弱,因而在样品Ni-N-OMC-2的高倍TEM图4(f)中未能观察到环绕于Ni纳米粒子周围的石墨层。

图4 样品的电镜图Fig.4TEM and HRTEM images of the samples OMC(a,b),Ni-N-OMC-1(c,d)and Ni-N-OMC-2(e,f)
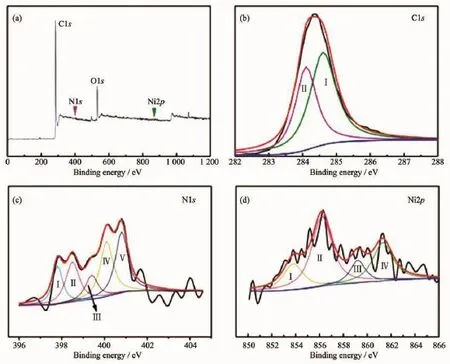
图5 Ni-N-OMC-1的XPS图谱:(a)全谱,(b)C1s,(c)N1s,(d)Ni2pFig.5XPS spectrum of Ni-N-OMC-1(a),C1s of Ni-N-OMC-1(b),N1s of Ni-N-OMC-1(c)and Ni2p of Ni-N-OMC-1(d)
采用XPS测试分析试样表面C,N和Ni元素的存在形式。样品Ni-N-OMC-1的XPS全谱图列于图5(a)中。图5(b)为样品的C1s谱图,C1s的拟合峰位于284.1和284.6 eV处,其中键能在284.1 eV处的Ⅰ峰来源于石墨化碳[20],键能位于284.6 eV的Ⅱ峰源于sp3杂化的C-C键的峰[21]。N1s的放大图谱列于图5(c)中,拟合的5个分峰分别位于397.8,398.5, 399.4,400.1和400.8 eV处。397.8 eV处的Ⅰ峰对应于sp3杂化与C结合的N原子[22],398.5 eV处的Ⅱ峰为吡啶N原子,即N代替了六元环中的C原子[23]。399.4和400.1 eV处的Ⅲ和Ⅳ峰对应于sp2杂化与C相结合的N原子[24-25],位于400.8 eV处的Ⅴ峰属于quaternary-N原子,即在四元环中与C相连的N原子[26]。通过上述分析可知,尿素与酚醛树脂在合成过程中发生交联反应,并在后续的热处理过程中与C原子形成稳定的化学键。N掺杂有助于改善碳载体的性能,研究发现,电子供体N在碳纳米管[27],石墨烯[28]或高石墨化程度的介孔碳[29]表面的掺杂提高了载体的π键强度,载体费米能级向导带移动,电导率明显增加。另一方面,含氮石墨化碳载体的π电子离域体系有利于金属离子的锚定,同时增加了离子间的静电排斥作用,有利于Pt的后续负载与分散[28],即使在载Pt量较高时也能防止Pt的团聚[30]。Ni2p图谱列于图5(d)中,拟合的4个分峰的位置分别为853.9,856.2,859.4和861.3 eV。其中859.4 eV处的Ⅲ峰来自于金属单质Ni[31],其余3个峰来自于NiO或Ni2O3[31-32],XPS测试在试样表面的探测深度为1~5 nm,由于表面的Ni单质在空气中易被氧化,因而在图5(d)中金属Ni的特征峰较弱。
2.2 试样载Pt后的结构与电化学性能分析
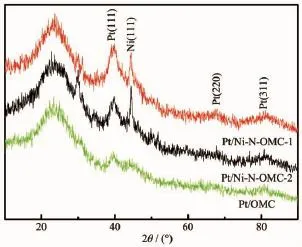
图6 载Pt样品的大角XRD图谱Fig.6XRD patterns of the samples with Pt loading
图6 为Pt/OMC,Pt/Ni-N-OMC-1,Pt/Ni-N-OMC-2复合材料的大角XRD图谱。其中39°,67°,81.3°处的衍射峰分别对应于Pt(111),Pt(220)和Pt(311)晶面,微波负载得到的Pt纳米粒子粒径较小,因而衍射峰强度较弱且峰型具有明显宽化现象[6]。44.5°处为金属Ni(111)晶面[14]的衍射峰。微波时的高温与微酸性环境可能导致金属Ni纳米微粒的再溶解,理论计算表明,金属Ni的密排面(111)晶面是Ni最稳定的表面[33],因而在微波过程中该晶面的Ni原子溶解缓慢,而晶面指数较高的(200)面和(220)面溶解迅速,因而大角XRD图谱中未出现(200)与(220)晶面的特征峰。为进一步观察Pt纳米粒子在碳载体表面的分散情况,对所制备的催化剂进行TEM和高分辨TEM分析。如图7(a)所示,只有少数Pt粒子负载于Pt/OMC表面。由表1可知,OMC的介孔比例最低,导致Pt离子前驱体溶液不易进入到碳材料内部进行反应;其次,OMC表面活性不足,还原生成的Pt纳米粒子和碳载体之间吸附作用弱,在后续清洗过程中易流失[34],因而Pt/OMC表面Pt负载量较少。由图(b)可见,试样Pt/Ni-N-OMC-1中,Pt颗粒密集且均匀地分布于载体表面,粒径小于5 nm,这是由于Ni-N的共改性改变了碳载体的理化性质,Ni以金属颗粒形态嵌于介孔碳骨架中,为Pt的负载提供活性位点;此外,金属Ni催化周围无定型碳向石墨态转变,石墨化碳与N孤对电子形成的π电子离域体系增强了Pt颗粒与碳载体的作用力,Pt颗粒的生长得到有效控制,提高了其分散度与稳定性;试样Pt/Ni-N-OMC-2中Ni担载量不足导致碳表面活性点缺失,如图(c)所示,其Pt负载量小于试样Pt/ Ni-N-OMC-1。

图7 载Pt样品的电镜图Fig.7TEM and HRTEM images of the samples with Pt loading
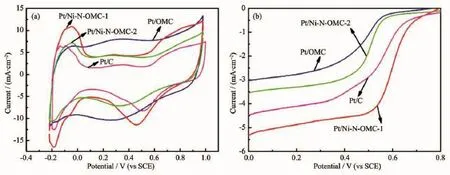
图8 催化剂的循环伏安曲线(a)和线性扫描伏安曲线(b)Fig.8CV curves(a)and LSV(b)curves of the catalysts

表2 催化剂的电化学分析结果Table 2Electrochemical parameters of the catalysts obtained from CV curves in 0.5 mol·L-1H2SO4
图8(a)为商业20%Pt/C,Pt/OMC,Pt/Ni-N-OMC-1,Pt/Ni-N-OMC-2在0.5 mol·L-1H2SO4溶液中的循环伏安(CV)曲线,扫描速度为20 mV·s-1。由图中可以观察到H2的氧化还原峰,电位正扫时CV曲线于-0.08 V附近出现氢的氧化峰,电位负扫时于-0.2 V左右出现氢的还原峰。电位负扫时相对电位在0.4~0.6 V之间的峰是氧或含氧粒子在Pt上的吸脱附引起的,由其相对位置和形状看,氧的吸脱附反应属于不可逆过程。对图8(a)进行分析所得电化学数据列于表2。OMC主要由无定形碳组成,表面活性低,导电性差,由图7(a)可知,只有少数Pt颗粒负载于OMC表面,因而Pt/OMC催化性能较弱,氢的氧化峰电流密度以及电化学活性面积分别为6.50 mA·cm-2,2.0 m2·g-1。试样Pt/Ni-N-OMC-1(10.92 mA· cm-2,138.53 m2·g-1)的电催化活性大于Pt/Ni-NOMC-2(7.29 mA·cm-2,53.28 m2·g-1),分析可能原因如下:试样Pt/Ni-N-OMC-1中金属Ni负载量大,结晶度高,具有较强的催化石墨化能力,碳载体导电率得到提高,有利于催化反应中电荷的快速传导;其次,Pt/Ni-N-OMC-1具有较大的比表面积(567 m2· g-1)以及最大的孔径(3.9 nm),高比表面积以及较大的孔径有利于提高传质速度,增大催化反应的三相界面,从而提高电催化反应的效率;由图7(b)可知,试样Pt/Ni-N-OMC-1中Pt担载量大,粒径小(不足5 nm)且分布均匀,Pt颗粒良好的负载与分散情况决定了试样Pt/Ni-N-OMC-1突出的催化性能,其氧化峰电流密度以及电化学活性面积分别为商业20% Pt/C(6.37 mA·cm-2,96.98 m2·g-1)的1.71倍和1.43倍。
图8(b)为氧还原反应的线性伏安(LSV)曲线。LSV曲线可以划分为3个明显的电位区间,以Pt/ Ni-N-OMC-1为例,0.8~0.68 V为动力学控制区,0.68~0.50 V为混合控制区,0.50~0 V为扩散控制区。如图8(b)所示,4组试样的氧还原起始电位以及极限电流密度分别为:商业20%Pt/C:0.64 V,4.49 mA·cm-2;Pt/OMC:0.55 V,3.03 mA·cm-2;Pt/Ni-NOMC-1:0.68 V,5.32 mA·cm-2;Pt/Ni-N-OMC-2:0.57 V,3.58 mA·cm-2。对比发现,Pt/Ni-N-OMC-1催化氧还原活性最强,由图7分析可知,该试样中Pt颗粒粒径较小且负载量大,因而与O2接触的催化位较多;其次,Pt/Ni-N-OMC-1具有良好的导电性,有利于反应过程中电子的快速传导;再次,Pt/Ni-N-OMC-1具有利于氧还原反应的结构特征,此外,Pt/Ni-NOMC-1中π电子离域体系与O2分子上的π电子相互作用,促进了氧分子在催化剂表面的有效吸附,有助于催化反应的发生。
3 结论
以F127为模板剂,间苯二酚甲醛树脂为碳源,原位添加尿素为氮源,氯化镍为镍源,分别采用均相法和两相法制备得到Ni-N-OMC-1,Ni-N-OMC-2纳米复合材料。Ni盐经碳热还原后主要以Ni微粒形式嵌入碳骨架中,原位催化无定形碳石墨化,为金属Pt沉积提供活性位点。N掺杂提高了石墨化有序介孔碳的电导率,增强了载体与Pt空轨道的相互作用,从而提高了Pt的分散度与稳定性。两相法制备Ni-N-OMC-2过程中,Ni2+与间苯二酚-甲醛-尿素共缩聚树脂配位作用较弱,Ni2+在静置时析出,导致Ni负载量偏低。采用均相法制备的Ni-N-OMC-1复合材料,Ni纳米粒子负载量大,结晶度较高,催化石墨化能力强。此外,该材料具有优异的结构特征,微波负载金属Pt后,显示出良好的催化性能,试样Pt/ Ni-N-OMC-1的氧还原极限电流密度为5.32 mA· cm-2,氢氧化电化学活性面积为138.53 m2·g-1,分别为商业20%Pt/C材料(4.49mA·cm-2,96.98 m2·g-1)的1.18倍和1.43倍。
[1]WANG Dao-Jun(王道军),WANG Tao(王涛),ZHOU Jian-Hua(周建华),et al.Chinese J.Inorg.Chem.(无机化学学报),2010,26(2):305-312
[2]Wang J C,Liu Q.J.Phys.Chem.C,2007,111(20):266-7272
[3]Zhao X C,Wang A,Yan J W,et al.Chem.Mater.,2010,22 (19):5463-5473
[4]Liang C D,Dudney N J,Howe J Y.Chem.Mater.,2009,21 (19):4724-4730
[5]Li Z L,Liu J H,Xia C G,et al.ACS Catal.,2013,3(11):2440-2448
[6]Datta K K R,Balasubramanian V V,Ariga K,et al.Chem. Eur.J.,2011,17(12):3390-3397
[7]Wickramaratne N P,Perera V S,Park B W,et al.Chem. Mater.,2013,25(14):2803-2811
[8]Lu A H,Li W C,Salabas E L,et al.Chem.Mater.,2006,18 (8):2086-2094
[9]Chen Z,Weng D,Sohn H,et al.RSC Adv.,2012,2(5):1755-1758
[10]TANG Jing(汤静),WANG Tao(王涛),HE Jian-Ping(何建平),et al.Acta Chim.Sinica(化学学报),2011,69(15):1751-1759
[11]Tang J,Wang T,Pan X C,et al.J.Phys.Chem.C,2013, 117(33):16896-16906
[12]Wang X Q,Liang C D,Dai S.Langmuir,2008,24(14):7500-7505
[13]Meng Y,Gu D,Zhang F Q,et al.Angew.Chem.,2005,117 (43):7215-7221
[14]Wei Z Q,Xia T D,Ma J.Mater.Charact.,2007,58(10):1019 -1024
[15]Jia N Q,Wang Z Y,Yang G F,et al.Electrochem.Commun., 2007,9(2):233-238
[16]Shao Y Y,Yin G P,Gao Y Z.J.Power Sources,2007,171 (2):558-566
[17]Yu X W,Ye S Y.J.Power Sources,2007,172(1):145-154
[18]Liu S H,Wu M T,Lai Y H,et al.Mater.Chem.,2011,21 (33):12489-12496
[19]Yao J Y,Li L X,Song H H,et al.Carbon,2009,47(2):436-444
[20]Li Q,Yang J P,Feng D,et al.Nano Res.,2010,3(9):632-642
[21]Moncoffre N,Hollinger G,Jaffrezic H,et al.Nucl.Instrum. Meth.B,1985,7-8(1):177-183
[22]Wang P F,Takeno T,Adachi K,et al.Appl.Surf.Sci., 2012,258(17):6576-6582
[23]Geng D S,Hu Y H,Li Y L,et al.Electrochem.Commun., 2012,22:65-68
[24]Delpeux S,Beguin F,Benoit R,et al.Eur.Polym.,1998,34 (7):905-915
[25]Lahaye J,Nansé G,Bagreev A,et al.Carbon,1999,37(4): 585-590
[26]Yang D S,Kim C,Song M Y,et al.J.Phys.Chem.C,2014, 118(30):16694-16702
[27]Shao Y Y,Sui J H,Yin G P,et al.Appl.Catal.B:Environ., 2008,79(1):89-99
[28]SHI Guo-Yu(史国玉),WANG Zong-Hua(王宗花),XIA Jian-Fei(夏建飞),et al.Acta Chim.Sinica(化学学报),2013,71 (02):227-233
[29]Liu R L,Wu D Q,Feng X L,et al.Angew.Chem.,2010, 122(14):2619-2623
[30]Yue B,Ma Y W,Tao H S,et al.J.Mater.Chem.,2008,18 (15):1747-1750
[31]Liu H,Guo R X,Liu Y,et al.Surf.Coat.Technol.,2012, 206(15):3350-3359
[32]Prieto P,Nistor V,Nouneh K,et al.Appl.Surf.Sci.,2012, 258(22):8807-8813
[33]WANG Bo(王博),ZHANG Jian-Min(张建民),LU Yan-Dong (路彦冬),et al.Acta Phys.Sin.(物理学报),2011,60(1):506-514
[34]Zhou J H,He J P,Ji Y J,et al.Electrochim.Acta,2007,52 (14):4691-4695
Synthesis and Electrocatalytic Performance of N-Doped Ordered Mesoporous Carbon-Ni Nanocomposite
PAN Xu-ChenTANG JingXUE Hai-RongGUO HuFAN Xiao-LiZHU Ze-TaoHE Jian-Ping*
(College of Material Science and Engineering,Nanjing University of Aeronautics and Astronautics,Nanjing 210016,China)
Highly ordered mesoporous carbon co-modified with Ni-N can be prepared via homogeneous phase route as well as dual-phase route,named as Ni-N-OMC-1 and Ni-N-OMC-2 respectively.Triblock copolymer Pluronic F127 were employed as the template agent,urea as the N precursor,NiCl2as the Ni source and resorcinol-formaldehyde resin as the carbon precursor.X-ray diffraction(XRD),Raman,and transmission electron microscope(TEM)showed that nickel particles dispersed in the carbon matrix in forms of metal nickel,in situ catalyzing the graphitization of amorphous carbon.X-ray photoelectron spectroscopy(XPS)revealed that urea existed in four different N species after heat treatment:sp3nitrogen atoms bonded to carbon atoms,pyridine-like N,sp2nitrogen atoms bonded to carbon atoms and quaternary-N atoms.The co-modification of nitrogen and nickel changed the physicochemical properties of carbon matrix,thus making for the loading and dispersing of Pt. Pt nanoparticles deposited on Ni-N-OMC-1 nanocompsite showed excellent electrocatalytic activity.The electrochemical active surface area of hydrogen oxidation was 138.53 m2·g-1and the limiting current density in ORR was 5.32 mA·cm-2,which indicated higher electrocatalytic ability than that of the commercial 20%Pt/C catalysts(4.49 mA·cm-2,96.98 m2·g-1).
homogeneous phase route;dual-phase route;ordered mesoporous carbon;co-modified with Ni-N;electrocatalytic activity
TB333
A
1001-4861(2015)02-0282-09
10.11862/CJIC.2015.039
2014-08-12。收修改稿日期:2014-11-05。
国家自然科学基金(No.51372115)资助项目。
*通讯联系人。E-mail:jianph@nuaa.edu.cn
