BeH2与小分子间双氢键的理论研究
2015-03-20冯璐
冯 璐
(辽宁石化职业技术学院,辽宁 锦州 121000)
BeH2与小分子间双氢键的理论研究
冯 璐
(辽宁石化职业技术学院,辽宁 锦州 121000)
本文选择BeH2…HY(Y=CH3,C2H3,C2H,CN,NC)体系进行系统研究,探讨体系内H…H距离与体系相互作用能之间的变化规律。对双氢键体系的系统的理论研究能为人们进一步揭示H…H相互作用的本质提供更多更详实的证据。
双氢键 从头计算 密度泛函理论 分子间作用能
双氢键的成键情况特殊,仍具有饱和性和方向性,一些双氢键的强度与常规氢键的强度相当,这使得双氢键能影响物质的结构、反映性及选择性,因而在催化和晶体工程等领域具有潜在的应用背景[1]。
本文对BeH2…HY(Y=CH3,C2H3,C2H,CN,NC)复合物在MP2/6-311++g(3df,2p)和B3LYP/6-311++g(3df,2p)水平进行几何构型优化,通过频率验证得到没有虚频的结构(如图1所示),计算该复合物体系的键能De、离解能D0和相互作用能Eint,并分析体系键能与H…H距离之间的变化规律。
1.计算方法
复合物体系分子间作用能(De)的计算公式为:
De=E(X—H…H—Y)-[E(XH)+E(HY)]
对键能De进行零点能ZPE(zero-pointvibrationalenergy)校正得到离解能D0。基函数重叠误差BSSE(BasisSetSuperpositionError)校正的相互作用能Eint是采用Boys和Bernardi提出的均衡校正法计算得到的。需要注意的是,本文中计算的键能De、离解能D0和相互作用能Eint都没有考虑形变能(deformationenergies),这是由于单独优化的单体结构与复合物中单体的结构其差别是很微小的[2]。本文相关计算均采用Gaussian03程序包完成。
H—Be—H…H—Y
图1BeH2…HY(Y=CH3,C2H3,
C2H,CN,NC)体系的优化几何结构
2.结果与讨论
表1列出了键能De,离解能D0和相互作用能Eint。文献中BeH2…HY体系的能量也列于表中。从表1中的数据我们看出,BeH2…CH4体系和BeH2…C2H4体系的键能很小,经过零点能校正后的能量甚至是负值,结合表1中的H…H间距离(大于2.4),都说明BeH2…CH4体系和BeH2…C2H4体系的相互作用非常弱,不属于双氢键相互作用,属于弱的范德华相互作用。例如BeH2…CH4体系采用MP2/6-311++g(3df,2p)方法计算出的键能De、离解能D0和相互作用能Eint分别为0.30kcal/mol、-0.10kcal/mol和0.20kcal/mol;相对应的H…H间距离为2.711。对于BeH2…C2H2、BeH2…HCN和BeH2…HNC体系,我们的计算结果与已有的文献数据非常接近。例如Sadlej等人[2]利用MP2/aug-cc-pVTZ方法计算出的BeH2…C2H2体系的键能约为1.12kcal/mol,而采用MP2/6-311++g(3df,2p)方法计算的结果约为1.24kcal/mol,两者之间的差距非常小,仅为0.12kcal/mol,这更进一步的说明了我们所采用的方法和基组对目前研究的体系的恰当的。
表1 键能De,离解能D0和相互作用能Eint
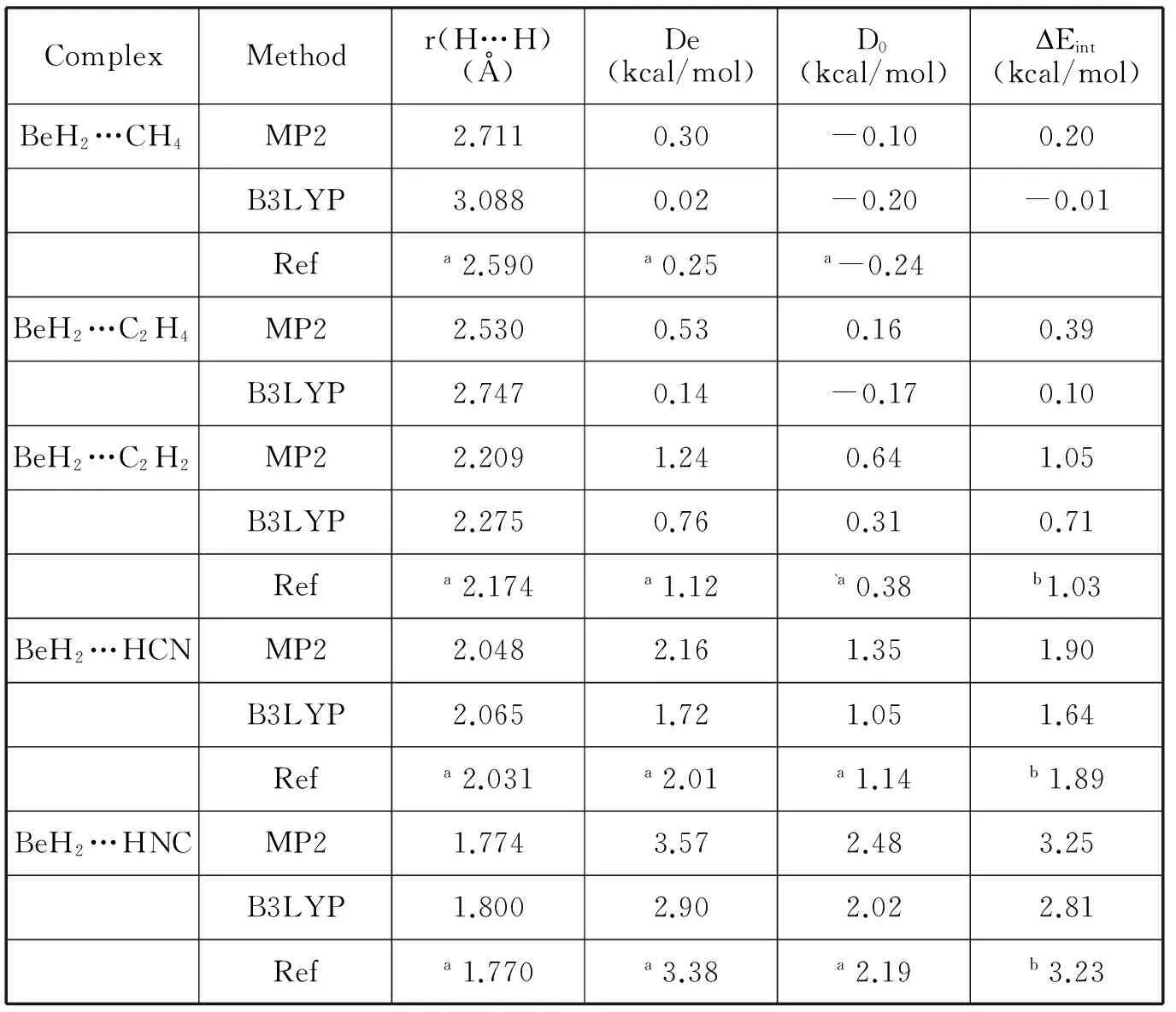
表1 键能De,离解能D0和相互作用能Eint
ComplexMethodr(H…H)(Å)De(kcal/mol)D0(kcal/mol)DEint(kcal/mol)BeH2…CH4MP22.7110.30-0.100.20B3LYP3.0880.02-0.20-0.01Refa2.590a0.25a-0.24BeH2…C2H4MP22.5300.530.160.39B3LYP2.7470.14-0.170.10BeH2…C2H2MP22.2091.240.641.05B3LYP2.2750.760.310.71Refa2.174a1.12`a0.38b1.03BeH2…HCNMP22.0482.161.351.90B3LYP2.0651.721.051.64Refa2.031a2.01a1.14b1.89BeH2…HNCMP21.7743.572.483.25B3LYP1.8002.902.022.81Refa1.770a3.38a2.19b3.23
注:aRef.2,bRef.3.BeH2…HY(Y=CH3,C2H3,C2H,CN,NC)体系的键能De、离解能D0和相互作用能Eint,表中包括文献中相应的相互作用能。
而且,值得注意的是Sadlej等人的计算中考虑了形变能,而在我们的计算中并为予以考虑,这是因为复合物与相应单体之间的差距是相当小的[2]。MP2/6-311++g(2d,2p)水平下计算出的BeH2…C2H2、BeH2…HCN和BeH2…HNC体系[3],经过BSSE校正后的相互作用能量分别为1.03kcal/mol、1.89kcal/mol和3.23kcal/mol,这非常接近我们采用MP2/6-311++g(3df,2p)方法,并经过BSSE校正后的能量Eint1.05kcal/mol、1.90kcal/mol和3.25kcal/mol。由表1可知能量的递增顺序为BeH2…CH4BeH2…C2H4BeH2…C2H2BeH2…HCNBeH2…HNC,这恰好与H…H间距离的变化趋势相反。BeH2…C2H2、BeH2…HCN和BeH2…HNC体系键能De的范围在14kcal/mol,这个能量变化区间接近于只有一个单氢键的中性复合物,例如水的二聚物[4]。如果我们从实验中测算氢键的能量[2],则BeH2…C2H2,BeH2…HCN和BeH2…HNC这三个双氢键体系可被视为典型的弱氢键相互作用。
采用相同6-311++g(3df,2p)基组,在B3LYP下计算的BeH2…HY(Y=CH3,C2H3,C2H,CN,NC)体系的相互作用能的变化规律与MP2方法下得到变化规律是相同的,但是,采用B3LYP方法计算出的相互作用能量都要比MP2方法计算出的小。例如,对于BeH2…C2H2体系,用MP2方法计算出的键能De为1.24kcal/mol,而采用B3LYP方法计算出的De为0.76kcal/mol,前者比后者能量高出0.48kcal/mol。这一结果说明,相较于MP2方法,B3LYP方法应用于讨论含有静电相互作用的双氢键体系是存在缺陷的。
3.结论
BeH2与HY(Y=CH3,C2H3,C2H,CN,NC)形成的复合物中BeH2…CH4和BeH2…C2H4体系存在弱的范德华相互作用;BeH2…C2H2、BeH2…HCN、BeH2…HNC体系的键能和H…H间距离都符合双氢键的定义,且形成的双氢键体系为直线型。体系能量的变化趋势为BeH2…CH4BeH2…C2H4BeH2…C2H2BeH2…HCNBeH2…HNC,H…H距离的变化规律与能量变化趋势相反,即键能越大H…H距离越小。
[1]CustelceanR,JacksonJE.Chem.Rev.2001,101,1963-1980.
[2]CybulskiH,TymińskaE,SadlejJ.ChemPhysChem.2006,7:629-639.
[3]AlkortaI,ZborowskiK,ElgueroJ.J.Phys.Chem.A2006,110:10279-10286.
[4]SchützM,BrdarskiS,WidmarkP-O,etc.J.Chem.Phys.1997,107:4597-4605.
