材料基因组计划与第一性原理高通量计算
2015-02-25范晓丽
范晓丽
(西北工业大学材料学院,陕西 西安 710072)

材料基因组计划与第一性原理高通量计算
范晓丽
(西北工业大学材料学院,陕西 西安 710072)
摘要:介绍了材料基因组计划的目标与核心思想,讨论了材料基因的来源与定义。虽然材料基因和计算材料都不是新事物,但两者均是材料基因组计划的核心要素,是加快新材料发展的关键。集成计算材料工程是材料基因组计划的基本要素,集合原子、微观、介观和宏观尺度计算工具的材料集成计算在新材料设计、工艺优化、环境响应方面发挥着重要作用。通过几个研究项目,介绍了第一性原理计算在新材料设计方面的应用,展示了高通量计算筛选新材料的强大功能。不仅如此,高通量计算结果和实验数据的结合将促进对材料物性的认识和材料基因组数据库的建设,为新材料设计提供有益信息。实施材料基因组计划,认识并建立材料结构与属性之间演化规律与新材料发现同等重要。此外,材料基因组计划还旨在变革材料研发理念与模式,在材料研发的全周期过程中采用交互、连续的流程模式,开发并集成计算工具、实验工具、数据信息三大基础构架模块。
关键词:材料基因;集成计算材料;第一性原理计算;高通量材料计算
1前言
为了使新材料从发现到市场应用的速度快两倍,2011年6月美国总统奥巴马宣布启动材料基因组计划(Materials Genome Initiative, MGI)[1],该计划是美国先进制造业伙伴关系的一部分,是振兴美国制造业的主要举措。
先进材料是科技创新、经济社会发展和提高全球竞争力的核心。材料基因组计划强调基础设施的兴建和开放合作方式的培育,以此加快先进材料的发展及市场化速度。材料基因组计划重点发展的先进材料包括清洁能源、人类福利和国家安全以及下一代生产力4个方面,它们是新生产力、新工具和新技术的基础。材料基因组计划的内容可用图1概括。中心是材料革新的基础构架,包括计算工具、实验工具、数据库三大模块,表示为相互交叉重叠的3个圆;3个圆的交叉重叠代表合作网络,整体体现独立开放的合作方式。三大模块整合集成并与产品设计框架无缝结合,将形成快速、整体的工程设计,使制造业更具竞争力。计算工具在三大模块中处于主导地位,是材料基因组计划的核心要素。图的外圈表示MGI实施的重点对象,即当前社会4个需求方面的先进材料。
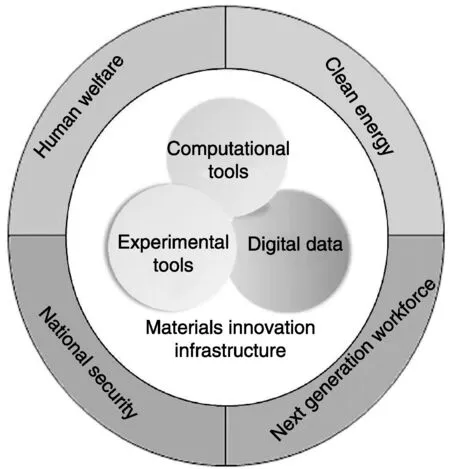
图1 材料基因组计划概述[1]Fig.1 Initiative overview[1]
在美国材料基因组计划宣布后,我国材料界也积极行动起来,陆续召开了多次相关研讨会,争取在新一轮材料革命性发展中占得先机。2014年9月21日,“2014新材料国际发展趋势高层论坛—材料基因组计划研究进展论坛”在西安举行,300余人的会场座无虚席。在20日的论坛大会报告中,中国版材料基因组计划项目组组长陈立泉院士介绍了我国开展MGI的重要性和紧迫性,并提出了开展中国版材料基因组计划的具体建议。材料基因组分论坛邀请了张统一院士、崔俊芝院士等9位报告人作了精彩报告,张统一院士的报告《上海大学材料基因组研究进展》,从材料研究的规划、组织和实施诸方面较为系统地介绍了上海大学材料基因组研究的进展以及刚刚成立的上海大学材料基因组研究院的建院思路。崔俊芝院士在其《新材料研发的集成化信息平台》报告中指出,集成化信息技术是支撑“材料基因工程”目标实现的关键技术之一。项晓东研究员的报告介绍了高通量组合材料实验与原位实时高通量组合材料实验技术的需求与发展现状。向勇、刘兴军、施思齐、陈亮、肖睿娟、刘利民等学者分别介绍了第一性原理、相图计算、相场模拟方法以及多尺度计算模拟在新型高温合金、锂离子电池材料、锂离子电池固体电解质材料筛选、光催化材料等先进功能材料的设计与研发方面的工作进展,阐明材料计算在新材料开发中的应用,以及计算和实验的相辅相成、相互促进的关系,进一步深化了材料设计的理念。
本文将介绍材料基因组计划的目标、核心思想、材料基因的概念与集成计算材料工程。基于计算材料在新材料研发模式中的主导地位及其在材料基因组计划实施过程中的核心作用,笔者结合自己的工作经验,重点探讨了第一性原理计算和高通量计算在新材料设计和筛选方面的应用。
2材料基因组计划的目标与核心思想
材料从发现到产品化一般经过7个步骤:发现-发展-优化-系统设计集成-证书-制造-应用。以今天的速度,一种新材料从发现到应用需要10~20 a的时间,MGI计划的目标是将材料从发现到市场的周期缩短一半。以往的材料研究大多采用尝试和改错的方法,即“炒菜”模式。MGI的做法是将传统的研发-产品的过程反转过来,从应用需求出发,倒推出符合相应功能材料的成分和结构。同时用交互、连续的流程替代传统试错法中的线性、分离流程,逐步由“经验指导实验”向“理论预测、实验验证”的新研究模式转变(如图2所示)。MGI将显著缩短学术研究和工业生产之间的间隙。
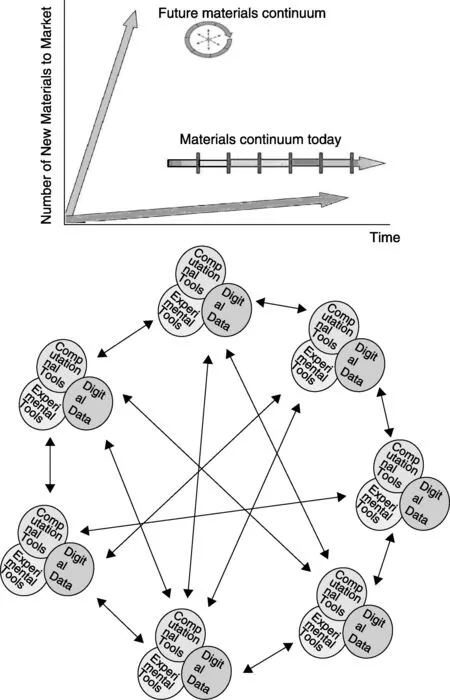
图2 材料全周期加速设计[1]Fig.2 Initiative acceleration of the materials continuum[1]
材料从发现到应用的7个步骤可以分为两个阶段,第一阶段是材料工程阶段,涉及材料的发现、优化、发展;第二阶段是产品工程阶段,涉及产品各部分的设计组合。材料发现-优化-发展需要10 a以上的时间,产品各部分的设计组合需要的时间少于2 a。因此,要实现材料从发现到市场化的周期缩短一半,主要工作应集中在第一阶段的材料工程上,大力提高材料发展的速度。
当前人类社会所需的先进材料涉及多组分和多相结构,成分结构复杂。没有精确的模型、充分的信息和数据交流,只用传统的经验方法来发展下一代材料将是一个艰难耗时的过程。为了探寻先进的材料设计能力以提升新材料发展的速度并降低成本,MGI强调材料革新基础设施的建设,这些设施包括计算工具、实验工具、数据信息,以及开放的合作关系和网络。这些模块经过开发及整合集成,应用于材料从发现到市场的整个周期中,全过程充分发挥合作关系和数据网络的作用(图2)。MGI材料研发模式不同于以往的研究模式,计算从辅助验证工具变为位于主导地位的指导工具。MGI强调了计算材料在开始的材料设计以及后期通往应用的每个步骤中的指导作用,彻底颠覆以往试错方法。实际上,计算机指导材料研究并不是一个新的话题,早在1991就有相关专著出版(《材料科学中的计算机模拟》)。MGI计划最直接的作用是使得依据计算能力以及相称的建模与模拟能力的增长创造新材料成为可能。
3材料基因与集成计算材料
材料基因(Materials Genome)这个词是美国滨州大学的刘梓葵教授在2002年创立材料基因组公司(www.materialsgeonom.com)时创造的[2],来源于相图计算方法CALPHAD (CALculation of PHAse Diagram, CALPHAD)[3]的成功应用和人类基因组计划所带来的灵感,后经材料基因组公司和白宫科学技术政策办公室双方同意,2011年6月,材料基因这个词被“材料基因组计划”采用。
人类基因是指人体中携带遗传信息的脱氧核糖核酸(Deoxyribonucleic Acid, DNA)片断,是控制人体性状的基本遗传单位。在材料基因组计划中材料基因并没有被精确定义:“基因组是一组以DNA为语言编码的信息,它描绘了有机体生长和发育的蓝图,当把基因组这个词应用在非生物领域时,意味着面向更大目标的基础性构造单元[1]”。人类基因组计划旨在通过对人类DNA中30亿个化学基元对(碱基对)的测序,发现人类基因组中所有基因的结构,建立DNA排序和人体机能性状之间的关系,实现分子水平上了解人类自身。材料基因组计划与人类基因组计划最大的相似点是两者都是从研究对象的最基本组份出发,试图了解“人”和“材料”,从而达到有目的地创造生物或者新材料的目的。
不同晶体结构单个相的吉布斯自由能和原子迁移率是温度、成分和压力的函数,Kaufman和Bernstein[3]认为多组元材料热力学的基本元素是单个相在完整的温度、压力和成分空间内的自由能函数。目前多组元材料的热力学和动力学数据库均以单个相作为基本组成模块。CALPHAD方法结合各组成相的热力学和动力学数据库以及物理性质,得到的单个相和相界面的性质可以作为相场法模拟的输入参数来模拟微结构演化,相场法模拟得到的材料微结构和相与相界面的性质进一步输入到有限元分析工具,以此来模拟材料的平衡状态以及对外界环境的响应。因此,单个相可以被认为是建立材料结构和性能之间关系规律、设计材料结构和性能的基本组成模块[4],但是,研究发现特定晶体结构相的内在构型,比如自旋方向和分布随着电场、磁场、温度和应力等外界条件而改变[5-6]。所以,科学家认为这些更加微观的内部构型(单个组态)[7-9]应作为单个相的基本组成模块,即被看作单个相的基因组[10]。原子尺度计算的电子结构和总能量相关的数据作为单个相内部构型的热力学和动力学数据,有助于确定实验难以精确测量的材料性质,如图3所示。
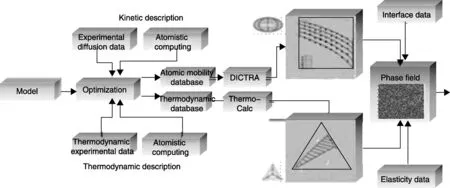
图3 CALPHAD计算微观结构的框架图[4]Fig.3 Frame diagram of CALPHAD calculating microscopic structure[4]
材料基因组计划目标实现的关键在于提升第一阶段材料发展的速度。材料的4个基本要素包括合成制备、结构、性质、服役性能,他们之间既相互独立又彼此关联,通常被表示为四面体的四个顶点。发展满足一定应用需求的材料,应从相应的服役性能出发,反式设计出材料的结构成分、合成方法和制备工艺。与反式工程设计过程相反,材料物性计算研究分为4个阶段:第一性原理计算、相图计算、相场模拟和有限元分析。由此获得热力学、动力学、晶体结构和缺陷相关的性质,以及对服役环境的响应。 前者致力于新材料的探索设计,从目标功能出发有目的地评估侯选材料组元,设计具备特定性质的材料。后者通过研究众多的材料组元,搜集有用的信息,寻找和建立材料原子结构-微观组织-宏观性能之间的联系。人类DNA的排列决定人体的主要机能性状,材料原子排列以及内部电子结构与外界环境共同决定材料的组织和性能。材料基因组计划中,反式工程设计理念和材料物性研究同等重要,两者有机结合才能实现材料基因组计划目标。
材料基因组计划提出建立更强的计算工具,创建更强的数据共享、管理和分析平台,这套全新的基础设施将为科学家和工程师们研究和创造新材料提供丰富的数据和信息。集成计算材料工程(Integrated Computational Materials Engineering, ICME)是材料基因组计划最基本的组成部分,其将通过计算工具所获得的材料信息与产品性能分析和制造工艺模拟集成在一起,目的在于设计新材料,或者在已有的材料上做改变以满足设计的需要。ICME的主要构建模块包括第一性原理、材料热力学与动力学、材料加工与性能的模拟工具。ICME通过材料模拟和开发设计之间的动态链接,尽可能发掘新材料的潜能,通过对制造工艺系统的每一部分进行优化,以提高产品零部件性能与质量稳定性、缩短开发周期、降低成本。集成计算材料工程涉及方法、应用、服务3个层面。微观、介观和宏观跨尺度计算方法层面的研究与直接针对需求目标应用层面的研究密切相关,不仅为其提供方法支撑,而且为计算材料科学自身发展做出贡献。服务层面包括材料数据库、材料计算程序、材料计算平台、相关资源的创建和提供。Hero-m项目(http:/www.hero-m.mse.kth.se/)与ICME和MGI密切相关,其研究内容如图4所示。该项目集合从原子到宏观各个尺度上的材料计算模拟方法与实验表征技术,开发材料设计过程中所需的ICME工具,并进一步将先进制造技术与材料实验和模拟融入到ICME工具中,开发材料选择、生产工艺、产品设计相结合的集成优化系统。
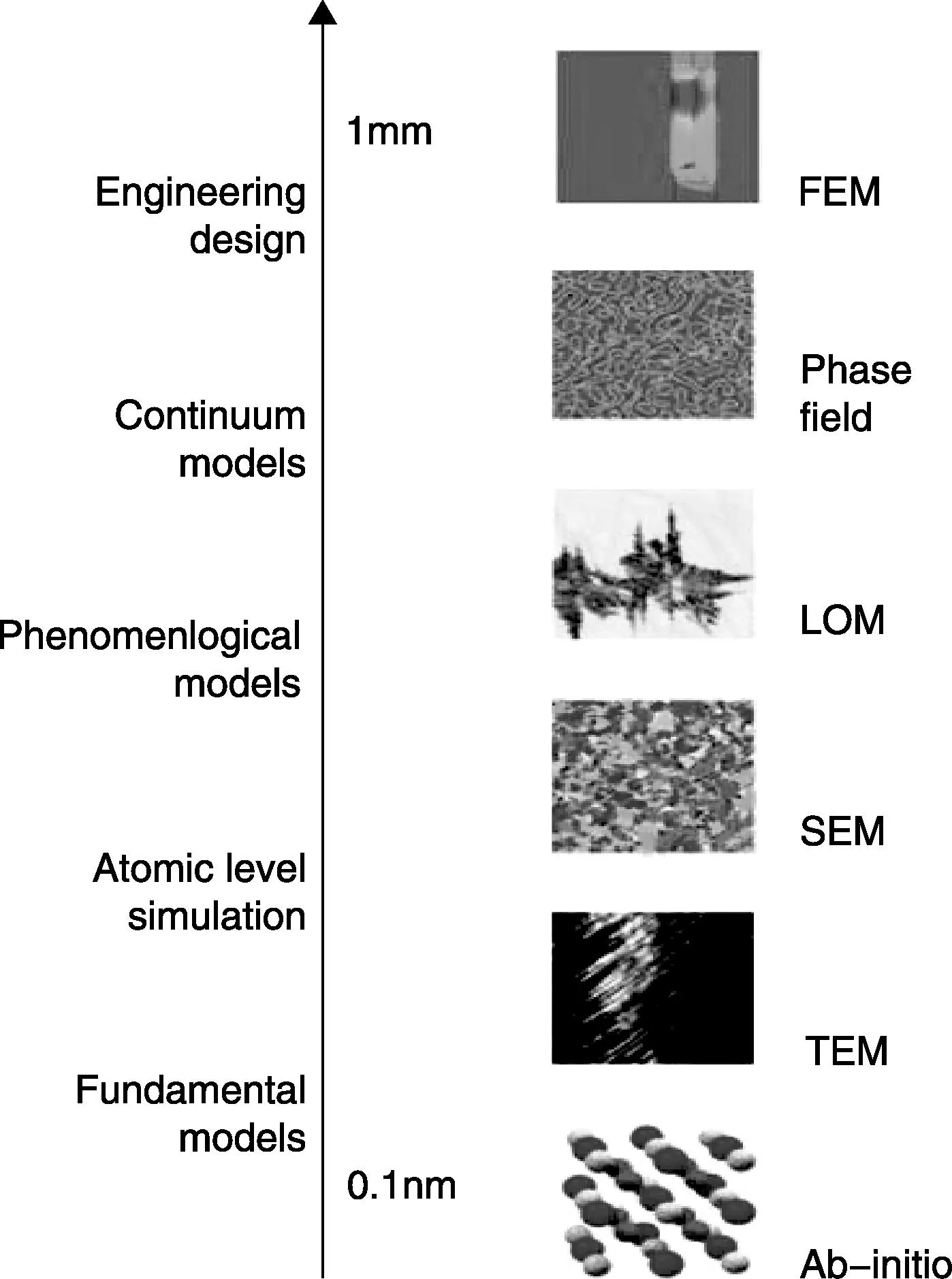
图4 Hero-m项目不同尺度材料工程设计Fig.4 Hero-m engineering design on different length scales
4第一性原理计算设计新材料
计算材料的发展对于材料基因组计划的实施至关重要,这一点在集成计算材料工程和Hero-m项目中均有体现。材料发现、优化、发展分别涉及原子、微观、介观尺度上的计算模拟。新材料发现旨在探索设计实验尚未制备或者自然界不存在的、结构未知的材料。按需设计是MGI的终极目标,设计特定性质材料的结构或者探索发现材料新的物相, 均与传统的材料性能优化和工艺模拟不同。基于经典理论的经验、半经验方法无法应用在该研究阶段。第一性原理方法基于量子力学原理,计算过程只需要所涉及的原子种类和位置坐标,此外不需要任何经验参数,该方法是新材料设计和探索的首选方法。
科学家们已经实现了在结构甚至化学计量比均未知,仅从材料的化学成分出发来预测材料新物相和设计特定性质的材料结构。美国纽约州立大学石溪分校的Oganov A主导开发的USPEX (Universal Structure Predictor: Evolutional Xtallography, USPEX)软件基于晶体结构预测方法[11-13],该软件能够基于材料的化学成分和给定的温度/压力,预测材料的稳定结构和一系列低能量介稳结构。此方法已推广到分子晶体、团簇、变成分结构、相变路径和基于力学和功能性质的晶体结构预测,部分研究成果见参考文献[14-16]。USPEX可以完全使用从头算方法处理晶胞含有6~40原子的体系。对于晶胞多于40个原子的体系,计算成本显著增大,但仍可以实现,需要借助USPEX中的其他方法或近似。对于100~200个原子的晶胞,使用经典力场方法,也可以得到很好的结果。同类的结构预测方法还有吉林大学超硬材料实验室的马琰铭主导开发的CALYPSO (Crystal structure AnaLYsis by Particle Swarm Optimization, CALYPSO) 程序包,该方法在晶体[17]、团簇[18]、二维层状材料[19]结构发现预测方面具有很高的成功率和收敛速度。
第一性原理方法能够准确计算原子结构下的各种电子结构和总能量相关的数据,预测热力学、动力学和力学性质,在考虑原子、电子尺度下振动和热电子熵的贡献后,能够预测有限温度下的性质。在为多组元材料设计服务的计算工具中,第一性原理计算作为第一阶段的计算工具参与多组元材料设计,使得单个相的内部微观构型成为多组元材料的基本组成模块,极大地增强了CALPHAD计算对多组元材料单个相性质的预测能力,如图2所示。
第一性原理计算方法在针对特定应用目标筛选组元材料方面同样取得了突出的成绩,这一点将在下一节说明。
5高通量计算筛选新材料
计算能力的提升以及计算模拟技术的发展,使得高通量计算搜索新材料成为可能。在真正的实验之前,我们可以采用高通量计算并根据特定需要来筛选自然界已有的或者不存在的,所有可能的复合物。同时,高通量计算和可用的实验数据相结合,建立结构和性质之间的联系,将为新材料的设计提供充分的数据信息,省掉“试”和“错”的过程。
最近,高通量计算已经开始应用于二次锂离子电池(Lithium Secondary Batteries, LSB) 材料的设计和发现中[20-24]。关系到二次锂离子电池性能的关键物理问题有:材料的电子结构和导电性,锂离子扩散动力学,电极、电解液表面、界面问题,电极材料的结构和相稳定性,离子植入热力学等。第一性原理计算的多功能性给以上绝大部分的物理问题的解决提供了有益的参考,重要的是,第一性原理计算的准确性足以预测二次锂离子电池的关键性能。首先,基于第一性原理的高通量计算方法可以计算数以千计的二次锂离子电池材料的性质,由此筛选出具备期望性质的材料。其次,结合第一性原理计算与微观、介观尺度上的高通量计算工具与数据挖掘技术,将帮助我们搞清楚一些重要的、控制二次锂离子电池材料性质的原理。中科院物理所陈立泉院士课题组依据关系到二次锂离子电池性能的相关物理问题,设计了一个筛选二次锂离子电池材料的高通量计算流程[23]。
清洁能源材料是材料基因组计划致力发展的4类先进材料之一。氢能是公认的清洁能源,其作为低碳和零碳能源正在脱颖而出。世界各国正在研究如何能大量而廉价的生产氢,利用太阳能分解水制氢是一个主要的研究方向。
光解水制氢涉及两步反应:
H++e-+*→H*,
2H*→H2+2*或者H*+H++e-→H2+*
制氢反应的第一步是氢吸附中间体的形成。热力学上,如果氢的吸附是强烈吸热过程,那么氢吸附中间体的形成将受阻碍;如果它是强放热过程,那么由吸附中间体形成H2则需要很大的能量导致反应难以进行。光解水制氢的关键在于找到一种高效廉价的催化剂,使得吸附中间体和H2的形成均容易发生。丹麦技术大学原子尺度材料物理中心的Noerskov J K课题组研究发现制氢反应的速度和反应自由能(ΔGH)之间存在一个火山模型[25],电流密度最大值出现在ΔGH=0eV处。 因此,氢吸附的吉布斯反应自由能成为判断金属材料[25-28]催化析氢反应活性的标准。该课题组以ΔGH为需求标准,成功将高通量计算应用在了制氢催化材料筛选上[28]。他们将催化活性标准、精确稳定性评估、基于700多个过渡金属表面合金的DFT计算数据有效地结合,形成评估程序,由此完成了大规模合金催化剂的组合筛选。计算结果如图5所示,显示Bi和Pt的合金表面是最有潜力的制氢催化材料之一。Noerskov J K课题组后来成功合成了BiPt合金表面,并证明了该材料的催化活性优于典型制氢催化剂Pt。最近,此类计算被用于评估过渡金属二硫化物的催化性能[29-30],计算结果与实验发现完全一致。
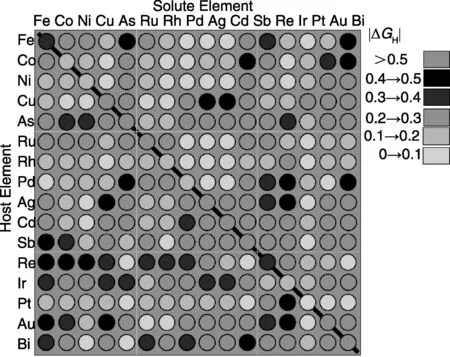
图5 高通量计算筛选256个纯金属和表面合金[28]Fig.5 Computational high-throughput screening for on 256 pure metals and surface alloys[28]
6结语
MGI实施以来,获得了美国学术界和工业界的积极响应,取得了较快的进展。我国也多次召开了各种类型的材料基因组研讨会,鉴于我国关键新材料的长期缺乏,历届与会专家一致认为我国必须启动中国版的“材料基因组计划”, 变革材料研发传统模式,加速国家关键新材料的研发和应用进程。材料基因组工程得到了各级政府的高度重视,参与的政府部门、学术机构、院校日趋增多,已经成立了多家材料基因组机构:上海市材料基因组研究院,材料基因工程北京市重点实验室,上海大学材料基因组研究院,西北工业大学材料基因组国际合作研究中心,北航集成计算材料工程中心等。
材料基因组计划将变革传统离散型的、经验试错法的研发模式,建立计算工具、实验工具、数据库相融合、协同创新的研发理念。计算在新材料研发模式中处于指导地位。计算工具建设的最终目标是使用仿真软件加速材料的研发部署,指导发现新材料、替代物理测试。计算工具当前的主要问题是还不具备多空间和时间尺度的仿真计算能力,同时各种软件分散、难以集成。目前应以开放的方式,加速计算工具的开发,特别是快速增长的第一性原理方法和先进模拟方法。材料集成计算工具在工业界已经发挥了作用,学术界和工业界的开放式互动交流将促进集成计算研究走向深入。随着计算能力、数据管理和材料科学与工程集成方法的发展,材料研发模式应转向开放共享和集成协作,同时提升文化氛围,使下一代材料人完全采纳这种方式,并使其进一步发展。
参考文献References
[1]United States of Office of Science and Technology Policy, National Science and Technology Council.MaterialsGenomeInitiativeforGlobalCompetitiveness. [EB/OL]. (2011-06-24). http://www.whitehouse.gov/sites/default/files/microsites/ostp/materials_genome_initiative-final.pdf.
[2]United States Patent and Trademark Office.TrademarkElectronicSearchSystem(TESS):MaterialsGenome. [EB/OL]. (2012-11).http://tess2.uspto.gov.
[3]Kaufman L, Bernstein H.ComputerCalculationofPhaseDiagram[M]. New York: Academic Press Inc., 1970:55-60.
[4]Campbell C E, Kattner U R, Liu Z K. File and Data Repositories for Next Generation CALPHAD[J].ScriptaMaterialia, 2014, 70(1): 7-11.
[5]Drautz R, Fähnle M. Parametrization of the Magnetic Energy at the Atomic Level[J].PhysRevB, 2005, 72(212 405):1-4.
[6]Lavrentiev M Y, Nguyen-Manh D, Dudarev S L. Magnetic Cluster Expansion Model for bcc-fcc Transitions in Fe and Fe-Cr Alloys[J].PhysRevB, 2010, 81(184 202): 1-6.
[7]Wang Y, Hector L G, Zhang H,etal. Thermodynamics of the Ce gamma-alpha Transition: Density-functional Study[J].PhysRevB, 2008, 78(104 113):1-9.
[8]Wang Y, Hector L G, Zhang H,etal. A Thermodynamic Framework for a System with Itinerant-electron Magnetism[J].JPhysCondensMatter, 2009, 21(326 003):1-7.
[9]Wang Y, Shang S L, Zhang H,etal. Thermodynamic Fluctuations in Magnetic States: Fe3Pt as a Prototype[J].PhilosMagLett, 2010, 90(12):851-859.
[10]Liu Zikui (刘梓葵). 关于材料基因组的基本观点及展望[J].ChineseScienceBulletin(科学通报), 2013, 58(35): 3 618-3 622.
[11]Lyakhov A O, Oganov A R, Valle M.CrystalStructurePredictionUsingEvolutionaryApproach.In:ModernMethodsofCrystalStructurePrediction[M]. Berlin: Wiley-VCH, 2010: 147-180.
[12]Oganov A R.CrystalStructurePrediction,aFormidableProblem.In:ModernMethodsofCrystalStructurePrediction[M]. Berlin: Wiley-VCH, 2010:6-21.
[13]Oganov A R, Ma Y, Lyakhov A O,etal.EvolutionaryCrystalStructurePredictionandNovelHigh-PressurePhases.In:High-pressureCrystallography[M]. Berlin: Springer, 2010:293-325.
[14]Liu Y, Oganov A R, Wang S,etal. Prediction of New Thermodynamically Stable Aluminum Oxides[J].SciRep, 2015, 5(9 518):1-10.
[15]Zhou X F, Oganov A R, Shao X,etal. Unexpected Reconstruction of the α-Boron (111) Surface[J].PhysRevLett, 2014, 113(17 6101) :1-5.
[16]Zhang W W, Oganov A R, Goncharov A F,etal. Unexpected Stoichiometries of Stable Sodium Chlorides[J].Science, 2013 (342):1 502-1 505
[17]Wang Y C, Lv J, Zhu L,etal. CALYPSO: A Method for Crystal Structure Prediction[J].ComputPhysCommun, 2012, 183(10): 2 063-2 070.
[18]Lv J, Wang Y C, Zhu L,etal. Particle-Swarm Structure Prediction on Clusters[J].JChemPhys, 2012, 137(084 104):1-8.
[19]Wang Y C, Miao M S, Liu J,etal. An Effective Structure Prediction Method for Layered Materials Based on 2D Particle Swarm Optimization Algorithm[J].JChemPhys, 2012, 137(224 108): 1-6.
[20]Hautier G, Jain A, Chen H L,etal. Novel Mixed Polyanions Lithium-ion Battery Cathode Materials Predicted by High-Throughput ab-initio Computations[J].JMaterChem, 2011 (21): 17 147-17 153.
[21]Hautier G, Jain A, Ong S P. Phosphates as Lithium-Ion Battery Cathodes: An Evaluation Based on High-Throughput ab initio Calculations[J].ChemMater, 2011, 23(15): 3 495-3 508.
[22]Jain A, Hautier G, Moore C J,etal. A High-Throughput Infrastructure for Density Functional Theory Calculations[J].ComputMaterSci, 2011, 50(8): 2 295-2 310.
[23]Ouyang Chuying, Chen Liquan. Physics towards Next Generation Li Secondary Batteries Materials:A Short Review from Computational Materials Design Perspective[J].SciChina-PhysMechAstron, 2013, 56(12):2 278-2 292.
[24]Gao Jian, Chu Geng, He Meng,etal. Screening Possible Solid Electrolytes by Calculating the Conduction Pathways Using Bond Valence Method[J].SciChina-PhysMechAstron, 2014, 57(8):1 526-1 535.
[25]Noerskov J K, Bligaard T, Logadottir A,etal. Trends in the Exchange Current for Hydrogen Evolution[J].JElectrochemSoc, 2005, 152(3):23-26.
[26]Hinnemann B, Moses P G, Bonde J,etal. Biomimetic Hydrogen Evolution: MoS2Nanoparticles as Catalyst for Hydrogen Evolution[J].JAmChemSoc, 2005, 127(15): 5 308-5 309.
[27]Choi W I, Wood B C, Schwegler E,etal. Site-Dependent Free Energy Barrier for Proton Reduction on MoS2Edges[J].JPhysChemC, 2013, 117(42): 21 772-21 777.
[28]Greeley J, Jaramillo T F, Bonde J,etal. Computational High-Throughput Screening of Electrocatalytic Materials for Hydrogen Evolution[J].NatMater, 2006 (5): 909-913.
[29]Fan X L, Yang Y, Xiao P,etal. Site-Specific Catalytic Activity in Exfoliated MoS2Single-layer Polytypes for Hydrogen Evolution: Basal Plane and Edges[J].JMaterChemA, 2014 (2): 20 545-20 551.
[30]Pan H. Metal Dichalcogenides Monolayers: Novel Catalysts for Electrochemical Hydrogen Production[J].SciRep, 2014, 4(5348):1-6.
(编辑惠琼)
Materials Genome Initiative and First-PrinciplesHigh-Throughput Computation
FAN Xiaoli
(School of Materials Science and Engineering, Northwestern Polytechnical University, Xi’an 710072, China)
Abstract:Both Materials Genome and computational materials are the key to speed up marketing new material although they both are not new. In the present article, the author introduces the goal and core concepts of MGI,and discusses the definition and understandings to Materials Genome. The integrated computational materials engineering (ICME) is the essential ingredient of MGI. Integrated computational materials combined the atomic, microscopic, mesoscopic and macroscopic scale computational tools play an important role from the initial discovery and optimizing to the environmental testing. Through a few projects, the applications of first-principles calculation in new materials design, and high-throughput computation in certain materials screening are presented. Combination of high-throughput computation and experiments will further people knowing materials, constructing database, and providing useful information for new materials design. For MGI, knowing the relation between the material structures and properties and designing new material are both important. Additionally, MGI aims to change the conventional R&D pattern, combining calculation, experiment and database, applying interactive and concurrent technological procedure in the whole process from discovery to applications.
Key words:Materials Genome; integrated computational materials; first-principles calculation; high-throughput computation
中图分类号:TB3
文献标识码:A
文章编号:1674-3962(2015)09-0689-07
DOI:10.7502/j.issn.1674-3962.2015.09.07
作者简介:范晓丽,女,1976年生,教授,博士生导师,Email: xlfan@nwpu.edu.cn
基金项目:国家自然科学基金资助项目(21273172)
收稿日期:2015-07-20
