益肺清化膏质量标准研究
2015-02-23周军,张茉,王杰
周 军,张 茉,王 杰
(天津市药品检验所,天津 300070)
益肺清化膏质量标准研究
周 军,张 茉,王 杰
(天津市药品检验所,天津 300070)
目的:提高益肺清化膏的质量标准。方法:采用薄层色谱法对益肺清化膏中的紫菀、拳参、甘草进行定性鉴别;采用高效液相色谱法测定黄芪中黄芪甲苷的含量,色谱柱为安捷伦ZORBAX SB-C18柱 (250 mm×4.6 mm,5 μm),流动相为乙腈-水(36∶64),柱温为40 ℃,流速为0.8 ml/min,蒸发光散射检测器。结果:薄层色谱中的特征斑点明显,重现性好,无干扰;黄芪甲苷在0.515~1.030 μg范围内线性关系良好, 平均回收率为102.39%,RSD为1.5%(n=6)。结论:本法专属性强、准确、快速,可有效控制产品的质量,保证了临床疗效。
益肺清化膏,紫菀,拳参,甘草,薄层色谱,黄芪甲苷,高效液相色谱法
益肺清化膏是天津中新药业集团股份有限公司达仁堂制药厂生产的品种,具有益气养阴、清热解毒、化痰止咳的功效,用于气阴两虚所致的气短、乏力、咳嗽、咯血、胸痛,也可用于晚期肺癌见上述症候者的辅助治疗。本品原质量标准收载于《国家食品药品监督管理局国家药品标准》WS3-460(Z-066)-2001(Z),标准中包括白花蛇舌草、党参、甘草、败酱草的薄层鉴别,黄芪甲苷的薄层扫描定量测定。现对益肺清化膏质量标准进行了提高、完善,在原标准基础上,对处方中甘草的TLC鉴别方法进行了修订,新增紫菀和拳参的TLC鉴别;并将原标准中黄芪甲苷的定量方法由薄层扫描法改为ELSD-HPLC法。修订后的质量标准,简化了试验提取步骤,提高了药品的质量控制指标,具有专属性强、准确性高的特点,现该标准已收录在《中国药典》2010年版第一增补本。
1 实验材料
1.1 仪器 岛津LC-20AD高效液相色谱仪,配有LC-20AD 泵、SIL-20自动进样器、Alltech ELSD-2000检测器、CTO-20AC柱温箱;LC-solution 色谱工作站。METTLER TOLEDO AG135 型及METTLER AE 100型电子天平(瑞士梅特勒-托利多集团生产);CAMAG薄层色谱成像系统(瑞士卡玛公司生产);薄层色谱用普通硅胶G板(烟台市化学工业研究所生产)。
1.2 试药 黄芪甲苷对照品(批号110781-200613,供含量测定用,纯度为99.8%)、没食子酸对照品(批号110831-200302)、紫菀对照药材(批号120956-200504)、甘草对照药材(批号121303-201003),均于中国药品生物制品检定所购置。益肺清化膏样品(天津中新药业集团股份有限公司达仁堂制药厂,批号5410016、5410021、5410022,规格为120 g/瓶)。实验中使用的处方药材均由天津达仁堂制药厂提供,由天津市药品检验所吴贵华副主任药师鉴定,均符合《中国药典》2010年版一部相关标准的要求。乙腈、甲醇为色谱纯,水为去离子,其他试剂均为分析纯。
2 方法与结果
2.1 薄层色谱鉴别
2.1.1 紫菀的TLC鉴别 取益肺清化膏10 g,加乙酸乙酯30 ml,超声处理30 min,滤过,滤液蒸干,残渣加乙酸乙酯1 ml使溶解,作为供试品溶液。另取紫菀对照药材0.5 g,照供试品溶液制备方法制成对照药材溶液。按处方配比取处方药材10 g(紫菀除外),按制法制得阴性样品,再按照供试品溶液制备方法处理,作为阴性样品溶液。按照《中国药典》2010年版一部TLC法试验,吸取上述三种溶液各4 μl,分别点于同一硅胶G薄层板上,以甲苯-乙酸乙酯-甲醇-甲酸(20∶10∶1∶1)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。结果供试品色谱中在与对照药材色谱相应的位置上显相同颜色的荧光斑点,阴性样品色谱在相应位置无干扰,见图1。

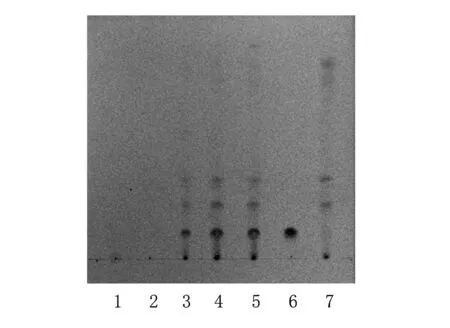
1.紫菀阴性样品溶液 2.紫菀对照药材 3.样品1(批号5410016) 4.样品2(批号5410021) 5.样品3(批号5410022) 6.没食子酸对照品 7.拳参阴性样品
2.1.2 拳参的TLC鉴别 取“2.1.1”项下的紫菀供试品溶液,作为供试品溶液。另取没食子酸对照品适量,加甲醇制成每1 ml含0.5 mg的溶液,作为对照品溶液。按处方配比取处方药材10 g(拳参除外),按制法制得阴性样品,再按照供试品溶液制备方法处理,作为阴性样品溶液。按照《中国药典》2010年版一部TLC法试验,吸取上述三种溶液各4 μl,分别点于同一硅胶G薄层板上,以甲苯-乙酸乙酯-甲醇-甲酸(20∶10∶1∶1),展开,取出,晾干,喷以5%三氯化铁乙醇溶液。供试品色谱中,在与对照品色谱相应的位置上显相同颜色的斑点,阴性样品色谱在相应位置无干扰,见图2。
1.紫菀阴性样品溶液 2.紫菀对照药材
2.1.3 甘草的TLC鉴别 取“3.2.2”项下30%乙醇洗脱液,蒸至无醇味,放冷,加乙酸乙酯提取3次,每次20 ml,合并乙酸乙酯提取液,蒸干,残渣加甲醇1 ml使溶解,作为供试品溶液。另取甘草对照药材0.2 g,加甲醇20 ml,超声处理30 min,滤过,滤液蒸干,残渣加甲醇1 ml使溶解,作为对照药材溶液。按处方配比取处方药材10 g(甘草除外),按制法制得阴性样品,再按照供试品溶液制备方法处理,作为阴性对照溶液。按照《中国药典》2010年版一部TLC法试验,取上述供试品溶液4~10 μl、对照药材溶液和阴性样品溶液各4 μl,分别点于同一硅胶G薄层板上,以乙酸乙酯-甲酸-冰醋酸-水(15∶1∶1∶2)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105 ℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上显相同颜色的斑点;置紫外灯(365 nm)下检视,在与对照药材色谱相应的位置上显相同颜色的荧光斑点,阴性样品色谱在相应位置无干扰,见图3。
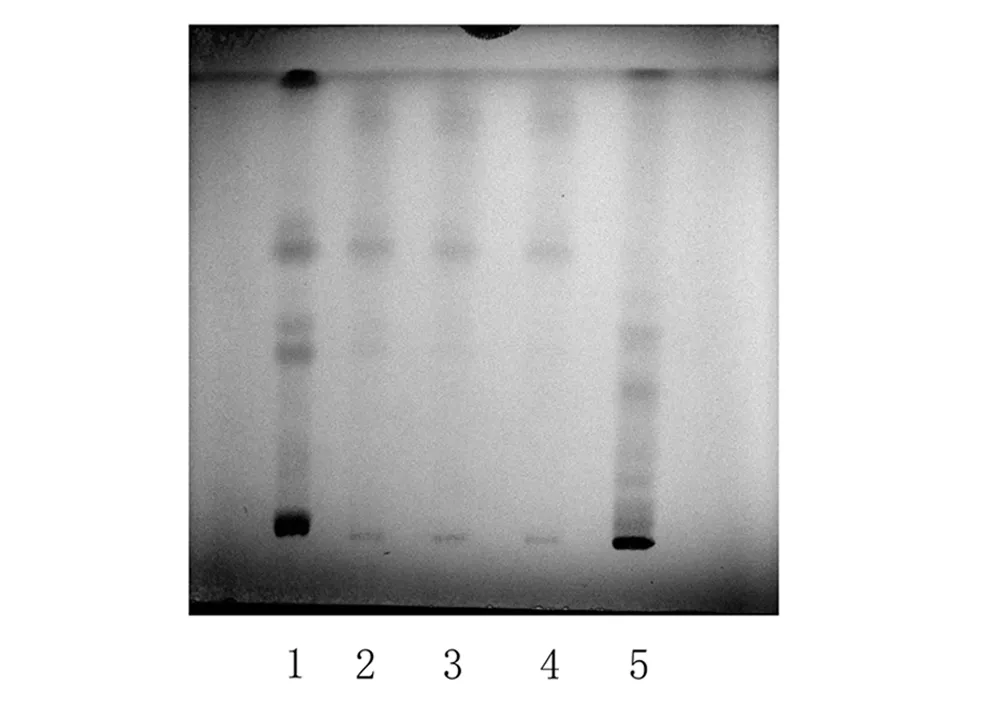
日光下
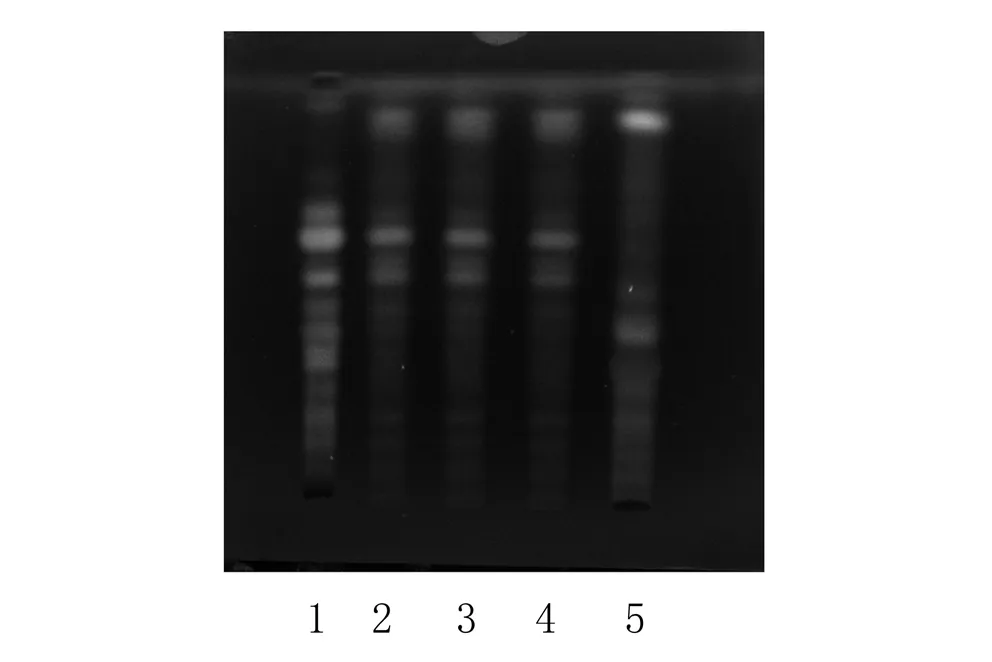
365 nm紫外光下
3 黄芪甲苷的测定
3.1 色谱条件 色谱柱:安捷伦ZORBAX SB-C18(250 mm×4.6 mm 5 μm);流动相为乙腈-水(36∶64);柱温为40 ℃:流速为0.8 ml/min。蒸发光散射检测器,漂移管温度100 ℃,气体流速3.0 L/min。理论板数按黄芪甲苷峰计算应不低于5 000。
3.2 溶液的制备
3.2.1 对照品溶液的制备 取黄芪甲苷对照品适量,精密称定,加甲醇制成每1 ml中含0.3 mg的溶液,即得。
3.2.2 供试品溶液的制备 取同一批号(5410022)样品4 g,置烧杯中,精密称定,加1%氢氧化钠水溶液30 ml使溶解,加于已处理好的D101大孔树脂柱上(内径1.5 cm,柱长20 cm),用1%氢氧化钠水溶液20 ml洗脱,再用水100 ml洗脱,继用30%乙醇50 ml洗脱,最后用90%乙醇75 ml洗脱,收集90%乙醇洗脱液,减压蒸至约1 ml,用50%甲醇定容至10 ml量瓶中,加50%甲醇至刻度,摇匀,即得。
3.2.3 阴性样品溶液的制备 按处方取除黄芪的各味药材,按制法制得阴性样品,再按“3.2.2”项下供试品溶液制备方法,制得阴性样品溶液。
3.2.4 专属性试验 分别精密吸取黄芪甲苷对照品溶液10和20 μl,供试品溶液和阴性对照溶液各20 μl,注入HPLC色谱仪,测定,结果阴性对照无干扰,见图4。
3.3 标准曲线的制备 取黄芪甲苷对照品适量,精密称定,加甲醇分别制成浓度为每1 ml中含黄芪甲苷0.025 75、0.051 5、0.103 0、0.206 0、0.309 0、0.412 0、0.515 0和1.030 mg的对照品溶液,分别精密吸取20 μl,注入液相色谱仪,按“3.1”项下色谱条件分析,分别测定各自峰面积,以对照品进样的量(μg)的对数值为横坐标,峰面积的对数值为纵坐标,求得回归方程:Y=5.398+1.638 4X(r=0.999 6)。结果表明黄芪甲苷在0.515~1.030 μg范围内线性良好。
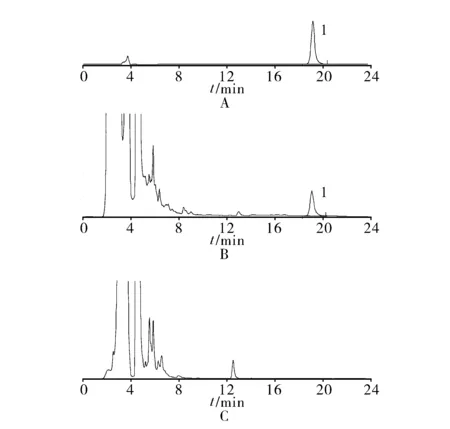
1.黄芪甲苷
3.4 精密度试验 取同一批号(5410022)样品4 g,精密称定,按照“3.2.2”项下供试品溶液制备操作,按“3.1”项下色谱条件分析,连续进样6次,测定峰面积,黄芪甲苷峰面积平均值为2 798 397.8,RSD为2.0%。
3.5 重现性试验 取同一批号(5410022)样品适量,共6份,取4 g,按照“3.2.2”项下供试品溶液制备操作,按“3.1”项下色谱条件分析。结果样品中黄芪甲苷平均含量为0.454 7 mg/g,RSD为1.5%。
3.6 稳定性试验 取本品(批号5410022)样品4 g,精密称定,按照“3.2.2”项下供试品溶液制备操作,按“3.1”项下色谱条件分析。分别在0、2、4、8、12和18 h进样,测定,黄芪甲苷峰面积平均值为2 749 823.8,RSD为3.5%。
3.7 回收率试验 取黄芪甲苷对照品适量,精密称定,加甲醇制成每1 ml含黄芪甲苷0.091 6 mg的对照品溶液,精密量取10 ml,共6份,分别置圆底烧瓶中,减压蒸干。取同一批号(5410022)样品2 g,置上述圆底烧瓶中,精密称定,按照“3.2.2”项下供试品溶液制备操作,制得供回收率用供试品溶液,按“3.1”项下色谱条件分析。计算回收率,结果黄芪甲苷平均回收率为102.39%,RSD为1.5%(n=6)。结果见表1。
3.8 样品测定 取3个批号的样品,按照“3.2.2”项下供试品溶液制备操作,按“3.1”项下色谱条件分析。结果见表2。
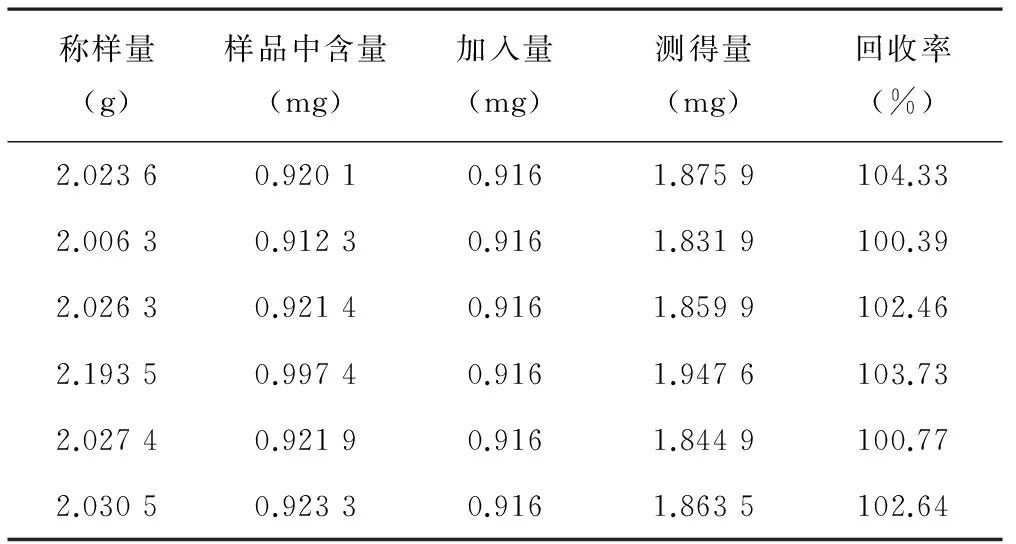
表1 回收率试验结果

表2 3批样品测定结果
4 讨论
4.1 益肺清化膏原标准中黄芪甲苷的定量方法为薄层扫描法,前处理方法极为烦琐,完成整个提取流程大约需要3 d的时间,为有效提高检验的效率,现对原方法进行修改,新起草的方法大约需要4~5 h即可完成样品的提取工作,极大地节省了试剂和时间,提高了工作效率。
4.2 黄芪甲苷的测定方法通常采用正丁醇萃取后加碱性溶剂洗涤,再将正丁醇提取液蒸干后,用甲醇溶解定容测定的方法[1-3]。黄芪中的皂苷类成分在碱性环境和加热的条件下,能够转化成黄芪甲苷,不能真实反应样品中黄芪的量[4-6]。本文采用大孔树脂吸附的方法既简化了步骤,又可以避免水解成分的产生,对控制药品质量有重要意义[7,8]。
4.3 提取方法的确定与考查
4.3.1 提取方式的考查 比较用 D101大孔树脂和正丁醇提取两种方法,结果正丁醇提取样品中黄芪甲苷的量低于过D101大孔树脂提取,因此采用过柱提取。
4.3.2 大孔树脂柱相关参数的考查 对D101大孔树脂柱的内径和柱长分别进行考查,结果确定内径为1.5 cm,柱长20 cm,样品中黄芪甲苷的量最高。
4.3.3 乙醇洗脱液浓度及体积的考查 经试验洗脱除去杂质时用30%乙醇50 ml效果最好;大孔树脂柱中吸附的黄芪甲苷用70%的乙醇即可洗脱下来,但90%乙醇比70%乙醇更容易蒸干,因此将70%乙醇调整为90%。
4.3.4 氢氧化钠溶液用量的考查 经试验用1%氢氧化钠水溶液30 ml可将样品完全溶解,再加于已处理好的D101大孔树脂柱上,继用1%氢氧化钠水溶液20 ml洗脱,可以减少氢氧化钠的用量,同时除去大部分的杂质干扰。
4.3.5 洗脱液流速的控制 用1%氢氧化钠溶液上柱时,为保证样品被充分吸附在大孔树脂柱上,流速控制在1 ml/min左右;用水和30%乙醇洗脱时,速度可稍快,流速为2~3 ml/min。用90%乙醇洗脱时,流速为1 ml/min。
1 中国药典[S].一部. 2010:283
2 夏广萍,刘鹏,韩英梅.不同处理方法和不同产地黄芪药材中黄芪甲苷的含量测定[J].中药材,2008,31(3):385-387
3 黄绳武,吴智慧,胡锡波,等.高效液相色谱-蒸发光散射检测法测定安心康滴丸中黄芪甲苷的含量[J].中国医院药学杂志,2007,27(11):1559-1561
4 赵陆华,吴孟华,单臻,等.高效液相色谱-蒸发光散射检测器法测定肾宝片及其中间体中黄芪甲苷的含量[J].药物分析杂志,2005,25(9):1009-1011
5 陈有根,辛敏通,杨滨,等.黄芪药材中黄芪甲苷含量测定方法的改进[J].中国新药杂志,2008,17(21):1857-1859
6 姜勇,金芳,鲍忠,等.不同来源黄芪药材中黄芪甲苷的定量分析[J].中国中药杂志,2006,31(11):930-933
7 郭宝林,张兰涛,刘京晶,等.蒙古黄芪不同种质黄芪甲苷含量比较[J].中国中药杂志,2009,34(24):3292-3294
8 刘玫,周晶,张庆伟,等.氨液水解法用于提高黄芪中黄芪甲苷含量的工艺研究[J].中国中药杂志,2008,33(6):635-638
2014-12-09
R927.11
A
1006-5687(2015)04-0010-04
