超高效合相色谱法同时测定复合维生素片中11种脂溶性维生素及其衍生物
2015-01-20周围王波刘倩倩杨盛鑫王丽婷
周围+王波+刘倩倩+杨盛鑫+王丽婷

摘 要 建立了超高效合相色谱法(Ultra performance convergence chromatography,UPC2)分离和测定复合维生素片中11种脂溶性维生素(A, D, E, K)及其衍生物的方法。超高效合相色谱(UPC2)技术集合超临界流体色谱(Supercritical fluid chromatography,SFC)和超高效液相色谱(Ultra performance liquid chromatography,UPLCTM)的技术优点,流动相以CO2为主体,乙腈为助溶剂梯度洗脱。选用Waters Acquity UPC2 HSS C18 SB色谱柱(100 mm × 3.0 mm 1.8 μm),流速1 mL/min,检测波长为284 nm。方法检出限在1.5~2.0 mg/L之间;VK1, VK2, VK3和VD3的线性范围分别为3~300 mg/L; VA、VA棕榈酸酯、VA甲酸、VE、VE醋酸酯、VE琥珀酸酯和VD2的线性范围分别为5~300 mg/L;加标回收率范围为97.31%~98.76%;相对标准偏差为0.41%~0.96%,可以满足复合维生素片中11种脂溶性维生素(A, D, E, K)及其衍生物的方法要求。
关键词 超高效合相色谱; 脂溶性维生素; 复合维生素片
1 引 言
脂溶性维生素包括维生素A, D, E, K及其衍生物,是维持机体正常生长、发育、代谢和机体生理功能所必需的物质。维生素A又称为视黄醇,只存在动物性食品中,具有维持视觉、生殖、细胞生长发育等生理功能[1];维生素E又称为生育酚,具有抗氧化、抗衰老、保护血管、促进生育的作用[2];维生素D为固醇类衍生物,能够促进钙在骨骼沉积,具抗佝偻病作用[3];维生素K又称抗出血维生素、凝血维生素,具有止血的作用[4]。如果缺乏或者摄入过量,就会引起代谢失衡,严重时会产生缺乏症疾病,甚至致人死亡[5]。但是脂溶性维生素在结构上极不稳定,在光照、氧气、高温及极端pH的条件下极易被氧化破坏[6],因此维生素A, E, D和K的同时测定目前仍然是检验技术的难点。
脂溶性维生素的有关测定方法主要有比色法[7]、荧光法[8]、分光光度法[9]、电化学法[10]、气相色谱法[11]、免疫分析法[12]、高效液相色谱法[13,14]。目前的国家卫生标准方法也仅限于对这4种维生素进行分别测定,这些方法的特异性不强,有时样品还要采用复杂的化学、物理或生物前处理,步骤繁杂、费时,而且干扰物质较多、要接触较多对人体有害的有机溶剂,容易造成维生素被破坏, 导致回收率下降,使测定结果偏低,不能用于多种物质的同时测定。有关同时测定多种脂溶性维生素的方法的文献很少,仅见于少数几篇液相色谱-质谱联用方法[15~17]。超高效合相色谱法(Ultra performance convergence chromatography,UPC2)除了具有传统超高效液相色谱的优点外,还可与超临界流体色谱技术结合,以超临界流体CO2为流动相主体,依靠流动相的溶剂化能力进行分离、分析的色谱过程[18,19]。加之超细(2 μm)填料技术,能够通过精确调节流动相强度、压力和温度,获得所需的系统分辨率和选择性,对待测物的保留和分离进行有效调控,具有操作温度低、有机溶剂使用量少、灵敏度高、重现性好、分析速度快等优点[20~22]。本研究采用超高效合相色谱法同时测定复合维生素片中的11种脂溶性维生素,在12 min内实现了11种脂溶性维生素(A, D, E, K)及其衍生物的分离。实验表明,本方法快速、准确、样品预处理简便、灵敏度高、重复性好、回收率较高、实用性强,为维生素定性与定量检测提供了一种高效可行的色谱检测方法。
2 材料与方法
2.1 仪器与试剂
超高效合相色谱仪(美国Waters公司),配有Waters EmpowerTM 3数据处理系统;3K30冷冻离心机(美国Sigma公司);MS3涡旋仪(德国IKA公司);移液枪(美国Thermo Electron公司,100~1000 μL、20~100 μL)。
维生素K1(纯度98.5%)、维生素K2(纯度99.5%)、维生素K3(纯度99.5%)、维生素A(纯度98.5%)、维生素A棕榈酸酯(纯度98%)、维生素A甲酸(纯度99%)、维生素E(纯度98.3%)、维生素E醋酸酯(纯度98.5%)、维生素E琥珀酸酯(纯度97.5%)、维生素D2(纯度98%)、维生素D3(纯度99.5%),德国Dr.Ehrenstorfer GmbH公司;CO2(纯度99.997%,兰州汇能公司);甲醇、乙腈、异丙醇、正己烷(色谱纯, 德国Merck KGaA公司);石油醚(分析纯,天津登峰公司);蒸馏水;复合维生素(美国安利纽崔莱公司)。
2.2 标准溶液配制
标准贮备液:精确称取每种维生素各0.030 g,用正己烷-异丙醇(8∶2, V/V)溶液溶解并定容至100 mL,配制成300 mg/L的标准储备液,4 ℃下冷藏待用(4 h后重新配制)。endprint
标准工作液:准确转移1000, 834, 500, 250, 100和25 μL标准贮备液,分别稀释为300, 250, 150, 75, 30, 7.5, 5.0, 1.0, 0.5和0.3 mg/L的标准工作液,4℃下冷藏待用(4 h后重新配制) 。
2.3 超高效合相色谱条件
Waters Acquity UPC2 HSS C18 SB色谱柱:(100 mm×3.0 mm, 1.8 μm),流动相:A为CO2, B为乙腈;梯度洗脱: 0~3 min, 95% A; 3~7 min, 85% A; 7~10 min, 60% A; 10~10.2 min, 60%~95% A; 10.2~12 min, 95% A。流速1 mL/min;进样量 1 μL;柱温50 ℃;检测波长284 nm;动态背压(ABPR): 1900 psi。
2.4 样品处理
复合维生素片剂:准确称取2.0 g研磨后的复合维生素片,于15 mL聚乙烯管中,加入10 mL异丙醇-正己烷(1∶1, V/V)溶液,用涡旋振荡器振摇10 min充分混匀后,高速冷冻离心机于4 ℃下以13000 r/min离心10 min, 取1 mL上清液,经0.22 μm滤膜过滤后,直接进样分离分析。
复合维生素胶囊:准确称取复合维生素胶囊内的油状液体1.0 g于15 mL聚乙烯管中,加入10 mL异丙醇-正己烷(1∶1, V/V)溶液,用涡旋振荡器振摇10 min充分混匀,经0.22 μm滤膜过滤后,直接进样分离分析。
3 结果与分析
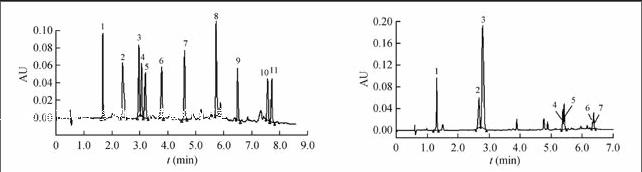
3.1 11种脂溶性维生素(A, D, E, K)及其衍生物的标准色谱图
使用HSS C18 SB柱时,在2.3节的色谱条件下分析浓度为75 mg/L的标准溶液,得到标准色谱图(图1)。
3.2 色谱分离条件的优化
3.2.1 色谱柱的选择 为了使11种脂溶性维生素(A, D, E, K)及其衍生物在较短时间内达到分离,并具有良好峰形,比较了Waters Acquity UPC2 BEH 2-EP (150 mm×2.1 mm i.d., 1.7 μm)和Waters Acquity UPC2 HSS C18 SB(100 mm×3.0 mm i.d., 1.8 μm)这两种色谱柱对11种脂溶性维生素(A, D, E, K)及其衍生物分离的影响。
使用BEH 2-EP柱(图2)时,11种脂溶性维生素(A, D, E, K)及其衍生物出峰不完全,其中VE琥珀酸酯、VE醋酸酯未出峰,而且VK1、VK2与VA棕榈酸酯出峰完全重叠,VD2和VD3色谱峰粘连在一起,分离效果不理想;当使用HSS C18 SB柱(图1)时,11种脂溶性维生素(A, D, E, K)及其衍生物在8 min内出峰完全,且峰形尖锐,相互之间无影响。因此,选择Waters Acquity UPC2 HSS C18 SB色谱柱进行分离。
3.2.2 流速的选择
超临界CO2作为流动相时,由于其黏度低,扩散系数高,使它在分离过程中具有较高的线速度,流速越大,被分离物质出峰时间越快,峰型尖锐,灵敏度增加,较小的流速会造成分析时间过长,峰展宽较大,影响灵敏度,故本实验通过使用超高效合相色谱,对亚2 μm填料的色谱柱的流速在0.6~1.2 mL/min 范围内进行优化。当流速为1.2 mL/min时,VE醋酸酯、VK1、VK2、VD2及VD3会因为流速过快而无法分离,或达不到分离要求;当流速为0.6 mL/min时,分离时间长达15 min,不能做到快速、高通量分析。为了保证较好的灵敏度、色谱柱压力以及尽可能与杂质分离,本实验最佳流速选择为1 mL/min。
3.2.3 助溶剂的选择
由于UPC2的流动相主要是CO2超临界流体,助溶剂的选择对于目标化合物的峰形及保留时间起着至关重要的作用,为了调整流动相对不同目标化合物的不同溶解性,通常加入甲醇、乙醇、乙腈、异丙醇、乙酸乙酯等助溶剂,有效改变目标化合物的峰形及保留时间。本试验分别选用了甲醇、乙醇和乙腈3种常用的不同极性的助溶剂,对相同浓度的11种脂溶性维生素(A, D, E, K)及其衍生物进行分离。结果表明,随着助溶剂极性的增加,流动相的溶解能力也相应得到增强,使得11种脂溶性维生素(A, D, E, K)及其衍生物的出峰时间提前。在使用甲醇为助溶剂时,由于甲醇较大的极性使得部分维生素(如维生素K1、维生素A甲酸)在流动相中的溶解能力降低而出现色谱峰分叉;使用极性较弱的乙醇作为助溶剂时,部分维生素(如VK1和VK2、VD2和VD3)不能很好地分离,且由于流动相的溶解能力使VA甲酸峰分叉。当选用助溶剂为乙腈时,11种脂溶性维生素(A, D, E, K)及其衍生物在较短的时间内达到较好的分离,并具有较好的峰型,因此,本实验选择乙腈为助溶剂。
3.2.4 动态背压(ABPR)的选择
超高效合相色谱中,动态背压(ABPR)是影响分离过程的重要因素之一。它主要作用是控制CO2在整个操作过程中的超临界流体状态。不同的动态背压下,CO2超临界流体对各种样品有着不同的溶解能力。当背压升高时,超临界流体密度会增大,溶剂化能力增强,柱压升高,分析物保留时间提前。本实验考察了背压在1800~2200 psi范围内CO2超临界流体对样品分离度的影响。结果表明,随着背压的增加,CO2超临界流体密度、黏度随之增加,柱压升高,当背压为2200 psi时,VE醋酸酯、VK1、VK2、VD2以及VD3分离度达不到分离要求。当背压为1800 psi时,因为CO2超临界流体溶剂化能力的降低,使得VD2和VD3色谱峰分叉,分析时间增加。通过对样品基质、保留时间、峰形及色谱柱压力的综合考虑,当背压为1900 psi时,11种脂溶性维生素(A, D, E, K)及其衍生物相互之间分离情况最好,保留时间较短、峰形对称,故本实验选择动态背压为1900 psi。endprint
3.2.5 色谱柱温度的选择 温度对CO2超临界流体影响也较大,一般在30~50℃之间进行选择。随着温度升高,超临界流体的黏度降低,溶剂化能力减小,出峰时间延长。随着温度降低,超临界流体黏度增加,溶剂化能力增强,出峰时间缩短;为了使样品中11种脂溶性维生素(A, D, E, K)及其衍生物得到较好的分离,本实验在保持其它色谱条件不变的前提下,在30~50℃ 范围内考察了柱温对目标物分离的影响。结果表明,随着温度的升高,11种脂溶性维生素(A, D, E, K)及其衍生物的保留时间逐渐增加,当温度为30 ℃时,虽然目标物均得到分离,但是柱压较高且重现性较差。当温度为50 ℃时,保证了脂溶性维生素(A, D, E, K)及其衍生物在分析过程中相互之间不干扰,并且具有较好的重现性。故本实验最佳分离温度选择50 ℃。
3.3 方法学考察
3.3.1 线性范围和灵敏度 将11种脂溶性维生素(A, D, E, K)及其衍生物系列标准工作液按2.3节色谱条件进行测定,绘制样品浓度(x,mg/L)与峰面积(y)标准曲线,进行线性回归分析,且当信噪比为3(S/N=3)时,方法对11种脂溶性维生素(A, D, E, K)及其衍生物的检出限(LOD)分析结果如表1所示。结果表明,本方法在表1所列的线性范围内,线性良好。
3.3.2 样品加标回收率和精密度 在空白样品中添加5, 10和50 mg/L的维生素标准溶液,涡旋混合,放置20 min,按2.4节进行前处理操作后,按2.3节进样测定,以浓度值计算加标回收率。为得到11种脂溶性维生素(A, D, E, K)及其衍生物的精密度,对每一添加浓度样品重复测定8次。各维生素的加标回收率在97.31%~98.76%之间,相对标准偏(RSD)为0.41%~0.96%。方法的回收率和重现性均较好。
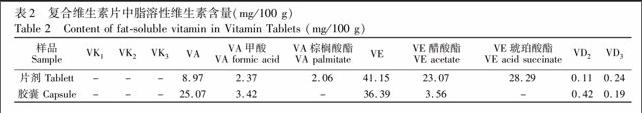
3.3.3 实际样品测定 在上述优化实验条件下,对维生素含量进行了分析,采用与标准保留时间相对照,外标法定量的方法测得两种复合维生素片(胶囊)中维生素的色谱图(图3),分析结果见表2。在本实验设定条件下样品及标样均能得到较好分离。
行检测,前处理过程简单,分析时间短,结果可靠,有效避免脂溶性维生素在长时间、复杂、繁琐的前处理及检测过程中的损失,导致定量不准确、实验过程误差较大的弊端,提高了脂溶性维检测的准确性。由于流动相为CO2超临界流体,对环境污染小,运行成本低。本方法为多种脂溶性维生素在食品中的同时检测分析提供了技术支持。超高效合相色谱(UPC2)将为未来的色谱分析提供新的方向。
References
1
Villamor E, Fawzi W W. Clin. Microbiol. Rev., 2005, 18(4): 446-464
2 ZHANG Xiao-Lei. Central South University of Forestry and Technology, 2012: 3-4
张晓雷. 中南林业科技大学, 2012: 3-4
3 HUANG Fang, WU Hui-Qin, HANG Yi-Ping. Physical Testing and Chemical Analysis Part B: Chemical Analgsis, 2011, 47(5): 577-579
黄 芳, 吴惠勤, 杭义萍. 理化检验-化学分册, 2011, 47(5): 577-579
4 Ahmed S, Kishikawa N, Nakashima K, Kuroda N. Anal. Chim. Acta, 2007, 591(2): 148-154
5 HUANG Cheng, LIN Sheng-Jun. Life Science Instruments, 2012, 10(4): 3-5
黄 诚, 林胜军. 生命科学仪器, 2012, 10(4): 3-5
6 CHENG Wan-Qin, WANG Jin, HUANG Li-Ying, ZHANG Shui-Feng, LI Hong-Yan, LIU Zhu, SHAO Liang-Liang, ZHANG Dong-Lei. Journal of Analytical Science, 2013, 29(1): 109-111
陈万勤, 王 瑾, 黄丽英, 张水锋, 李红艳, 刘 柱, 邵亮亮, 张东雷. 分析科学学报, 2013, 29(1): 109-111
7 LI Shu-Guo, XUE Wen-Tong, CHEN Hui, LI Xue-Mei. Cereals & Oils, 2006, 10: 26-29
李书国, 薛文通, 陈 辉, 李雪梅. 食品与油脂, 2006, 10: 26-29
8 Feng S, Gao F, Chen Z, Grant E, Kitts D D, Wang S, Lu X. J. Agric. Food Chem., 2013, 61(44): 10467-10475
9 LIU Shu-Heng. Chinese Journal of Spectroscopy Laboratory, 2010, 27(5): 890-892endprint
刘树恒. 光谱实验室, 2010, 27(5): 890-892
10 YI Shu-Tao, XUE Wen-Tong, ZHANG Ze-Jun, HUO Ting. Science and Technology of Food Industry, 2008, 29(12): 233-235
尹淑涛, 薛文通, 张泽俊, 霍 婷. 食品工业科技, 2008, 29(12): 233-235
11 SHAO Xiao-Fen, ZHANG Yun-Hui. Chinese Journal of Analysis Laboratory, 1994, 13(5): 61-62
邵晓芬, 张韵惠. 分析试验室, 1994, 13(5): 61-62
12 WANG Yu-Tang, CHI Tao, HU Ben-Tao, TAN Ju-Zhong, LIU Ning. Journal of Chinese Institute of Food Science and Technology, 2013, 13(12): 222-225
王玉堂, 池 涛, 胡本涛, 谭居中, 刘 宁. 中国食品学报, 2013, 13(12): 222-225
13 GB 5413.9-2010, Determination of Vitamin A, D, E in Foods for Infants and Young Children Milk and Milk Products. National Standards of the People′s Republic of China.
食品安全国家标准婴幼儿食品和乳品中维生素 A、D、E 的测定. 中华人民共和国国家标准. GB 5413.9-2010
14 Chavez-Servin JL, Castellote AI, López-Sabater M C. J. Chromatogr. A, 2006, 1122(1-2): 138-143
15 DONG Ai-Jun, CUI Yan-Hua, YANG Xin. Journal of Instrumental Analysis, 2011, 30(8): 887-891
董爱军, 崔艳华, 杨 鑫. 分析测试学报, 2011, 30(8): 887-891
16 QIU Li-Qun, GAO Ai-Ying, ZHANG Hao, JING Sheng. Guangzhou Chemical Industry, 2010, 38(10): 151-153
裘立群, 高艾英, 张 昊, 井 升. 广州化工, 2010, 38(10): 151-153
17 DUAN Yu-Lin, SU Jun, WU Xue, ZHANG Shao-Mei, WEN Tao, LIANG Meng-Yu, CHEN Xiao-Cong, MENG Yong, ZHOU Man, ZONG Yu-Ying. China Dairy Industry, 2012, 40(5): 58-60
段玉林, 苏 骏, 吴 雪, 张少梅, 温 韬, 梁梦宇, 陈小聪, 蒙 泳, 周 曼, 宗玉英. 中国乳品工业, 2012, 40(5): 58-60
18 LI Zhong-Hao, WU Shuai-Bin, LIU Shan-Shan, FAN Zi-Yan, YANG Fei, BIAN
Zhao-Yang, TANG Gang-Ling, CHEN Zai-Gen, HU Qing-Yuan. Chinese J. Anal. Chem., 2013, 41(12): 1817-1824
李中皓, 吴帅宾, 刘珊珊, 范子彦, 杨 飞, 边照阳, 唐纲岭, 陈再根, 胡清源. 分析化学, 2013, 41(12): 1817-1824
19 XU Yong-Wei, SUN Qing-Long, HUANG Jing, TAN Xiao-Jie. Modern Instruments, 2012, 18(5): 45-48
徐永威, 孙庆龙, 黄 静, 谭晓杰. 现代仪器, 2012, 18(5): 45-48
20 Gourmel C, Grand-Guillaume Perrenoud A, Waller L, Reqinato E, Veme J, Dulery B, Veuthey J L, Rudaz S, Schappler J, Guillarme D. J. Chromatogr. A, 2013, 1282: 172-177
21 Grand-Guillaume Perrenoud A, Veuthey J L, Guillarme D. J. Chromatogr. A, 2012, 1266: 158-167
22 Grand-Guillaume Perrenoud A, Boccard J, Veuthey J L. J. Chromatogr. A, 2012, 1262: 205-213endprint
Determination of 11 Fat-soluble Vitamins (A, D, E, K) and
Their Derivatives in Vitamin Tablets by Ultra Performance
Convergence Chromatography
ZHOU Wei*1,2,3, WANG Bo1, LIU Qian-Qian2, YANG Sheng-Xin3, WANG Li-Ting3
1(Central Laboratory of Technical Center of Gansu Entry-Exit Inspection and Quarantine Bureau,Lanzhou 730000, China)
2(College of Food Science and Engineering, Gansu Agricultural University, Lanzhou 730070, China)
3(College of Geography and Environment Science, Northwest Normal University, Lanzhou 730070, China)
Abstract A new method was developed for the determination of 11 fat-soluble vitamins (A, D, E and K) and its derivatives in vitamin tablets by ultra performance convergence chromatography (UPC2). The mobile phase was the mixture of supercritical CO2 and acetonitrile at a flow rate of 1 mL/min. The separation was carried out on the Waters Acquity UPC2 HSS C18 SB 100 mm× 3.0 mm i.d., 1.8 μm column. The UV detector was set at a wavelength of 284 nm. The limits of detection (LOD) were 1.5-2.0 mg/L, and the calibration linear for VK1, VK2, VK3 and VB3 was 3-300 mg/L, linear for VA, VA palmitate, VA formic acid, VE, VE acetate, VD2 and VD3 was 5-300 mg/L, respectively. Its spiked recoveries were 97.31%-98.76%, and the relative standard deviations (RSDs) were 0.41%-0.96%. The method is applicable for the determination of fat-soluble vitamins (A, D, E and K) and Their derivatives in vitamin tablets.
Keywords Ultra performance convergence chromatography; Fat-soluble Vitamin; Vitamin tablets
(Received 23 May 2014; accepted 28 September 2014)endprint
