嗜热共生杆菌内消旋-2,6-二氨基庚二酸脱氢酶中Y76对辅酶偏好性影响
2015-01-02赵雷明刘卫东陈曦王敏冯进辉吴洽庆朱敦明
赵雷明,刘卫东,陈曦,王敏,冯进辉,吴洽庆,朱敦明
嗜热共生杆菌内消旋-2,6-二氨基庚二酸脱氢酶中Y76对辅酶偏好性影响
赵雷明1,2,刘卫东2,陈曦2,王敏1,冯进辉2,吴洽庆2,朱敦明2
1天津科技大学生物工程学院 工业发酵微生物教育部重点实验室,天津 300457 2中国科学院天津工业生物技术研究所 工业酶国家工程实验室 天津市生物催化技术工程中心,天津 300308

赵雷明, 刘卫东, 陈曦, 等. 嗜热共生杆菌内消旋-2,6-二氨基庚二酸脱氢酶中Y76对辅酶偏好性影响. 生物工程学报, 2015, 31(7): 1108–1118.Zhao LM, Liu WD, Chen X, et al. Effect of residue Y76 on co-enzyme specificity of meso-diaminopimelate dehydrogenase from Symbiobacterium thermophilum. Chin J Biotech, 2015, 31(7): 1108–1118.
辅酶NAD(H) 相比NADP(H) 有稳定性好、价格低廉及更广的辅酶循环方法等优势,因此在实际应用中常需将NADP(H) 依赖型的脱氢酶改造成为NAD(H) 依赖型的。来源于嗜热共生杆菌的NADP(H) 依赖型内消旋-2,6-二氨基庚二酸脱氢酶 (-2,6-diaminopimelate dehydrogenase,StDAPDH) 及其突变体酶是催化还原氨化合成D-氨基酸的优良催化剂,本研究试图改变其辅酶偏好性,增强其应用优势。对其晶体结构分析可知,氨基酸残基Y76距离腺嘌呤较近,R35及R36和辅酶上磷酸基团有直接相互作用。依氨基酸侧链基团性质对Y76进行了定点突变,发现不同突变子对两种辅酶的偏好性都发生了变化;对与磷酸基团直接作用的R35、R36进行的双突变R35S/R36V,导致酶对NADP+的催化活力降低;将R35S/R36V和部分Y76突变进行了组合,发现三突变组合以NAD+为辅酶时的活力均大于以NADP+为辅酶的活力,实现了辅酶偏好性转变。这些研究工作为进一步实现StDAPDH的辅酶偏好性完全转变提供依据。
氨基酸脱氢酶,内消旋-2,6-二氨基庚二酸脱氢酶,辅酶偏好性,突变
氨基酸脱氢酶是以氨基酸分子中-CH-NH2为氢供体,以NAD(P)+为受体的一类氧化还原酶,有严格的手性选择性。野生型的氨基酸脱氢酶多为L-选择性[1],利用酶催化可逆反应的特点,这类酶被成功用于光学纯L-氨基酸的生物合成;目前已知的D-氨基酸脱氢酶多为膜蛋 白[2-8],仅有内消旋-2,6-二氨基庚二酸脱氢酶 (-2,6-D-diaminopimelate dehydrogenase) (DAPDH) (EC 1.4.1.16) 家族及它们的突变体酶可用来进行D-氨基酸的生物合成[9-12]。内消 旋-2,6-二氨基庚二酸脱氢酶 (DAPDH) 存在于赖氨酸合成途径中,是一类NADP+-依赖型的氧化还原酶,它催化可逆的内消旋-二氨基庚二酸(-diaminopimelate,-DAP) 的D-手性碳上的氨基氧化脱氨,生成L-2-氨基-6-酮基庚二酸,反应方程式如图1所示[13]。我们从嗜热共生杆菌中获得一个内消旋-2,6-二氨基庚二酸脱氢酶 (StDAPDH)[14],该酶可被直接用来还原氨化合成光学纯D-丙氨酸,我们对其进行改造扩展了底物谱范围[15],而且还获得了它的X-射线晶体结构[16],为对StDAPDH开展深入研究奠定了基础。
已报道的DAPDH家族野生型以及突变子酶均偏好于NADP(H)[9-12],到目前为止尚无该家族酶辅酶偏好性改造的报道。辅酶NAD(H)和NADP(H) 相比而言,NAD(H) 有稳定性好、价格低廉及更广的辅酶循环再生方法等优 势[17],在实际生产应用上,多倾向于使用NAD(H) 依赖型的脱氢酶。对DAPDH进行辅酶专一性改造,可以为DAPDH潜在的工业化应用提供支撑。

图1 内消旋-二氨基庚二酸脱氢酶催化的反应[13]
辅酶NAD(H) 和NADP(H) 的主要区别在于2′位上的腺嘌呤上,NADP(H) 中该位置比NAD(H) 多连接了一个磷酸基团,而且该位置和辅酶在催化反应中提供质子的C4的位置较远。在辅酶偏好性改造中,常常可以通过改造和辅酶磷酸基团结合的氨基酸残基如精氨酸 (R) 及赖氨酸 (K) 等来达到目的[18-30](表1)。
除了和磷酸基团相关的基团外,Crobu等[31]以及Baroni等[32]在改造铁氧化还原蛋白NADP+还原酶时发现,临近辅酶腺嘌呤的氨基酸残基 (H286及Y258) 对辅酶偏好性也有影响,而且他们获得的仅仅Y258位单突变就能使辅酶偏好性完全翻转过来。
通过结构分析比较发现,StDAPDH结构中和辅酶腺嘌呤接近并可能影响辅酶偏好性的氨基酸为Y76,与辅酶NADP+上磷酸基团有直接作用的基团为R35和R36。对于StDAPDH中Y76对辅酶偏好性是否有影响进行了突变摸索,此外,还对和与辅酶NADP+上磷酸基团有直接作用的R35/R36位也进行了突变尝试,并和Y76的部分突变子进行了组合突变测试。本文报道相关的研究结果。

表1 NAD(P)(H) 辅酶改造研究的总结
1 材料与方法
1.1 材料
1.1.1 菌株和质粒
实验中所用感受态细胞大肠杆菌Top10以及BL21 (DE3) 均购自北京全式金生物技术有限公司;突变起始质粒pET32a-为本实验室保存。
1.1.2 试剂
实验中所用KOD DNA聚合酶、PCR相关KOD plus缓冲液、MgSO4、dNTPs购自日本TOYOBO公司;Fast-Digest限制性内切酶购自Fermentas公司;辅酶NADP(H) 以及NAD(H) 均购自Codexis公司;文中所用底物以及其他试剂均购自Alfa Aesar公司。质粒提取试剂盒、核酸回收试剂盒均购自北京全式金生物技术有限公司。实验中所构建质粒均由金唯智生物科技有限公司进行序列测定。
1.1.3 培养基
文中所用培养基均为LB培养基,其中固体培养基中含1%琼脂粉。
1.1.4 主要仪器
酶标仪为MD公司Spectramax M2;蛋白纯化仪为ÄKTA purifier 10;所用层析柱以及填料均购自GE公司;APV2000高压匀浆破碎仪购自APV公司;RC6+高速冷冻离心机购自Thermo公司;离心机为Eppendorf公司5430R;MJ Mini PCR仪;核酸、蛋白电泳系统以及Gel DocTMXR+凝胶成像系统均购自Bio-RAD公司;SS-325高压灭菌锅购自日本Tomy Kogyo公司。
1.2 方法
1.2.1 定点突变
根据[14]基因序列和拟突变的位点设计相应突变引物 (表2),所有突变都以含突变前基因的质粒为模板,按照QuickChangeTM试剂盒操作说明进行相应突变。PCR过程:95 ℃预变性5 min,94 ℃变性30 s,60 ℃退火30 s,68 ℃延伸7 min,其中第二步至第四步循环20次,之后于68 ℃延伸20 min。PCR产物进行电泳确证。对扩增出来的目的片段进行切胶回收后,使用Ⅰ于30 ℃消化2 h,将消化产物转化至大肠杆菌Top10感受态细胞中,涂布至含氨苄青霉素的LB固体培养基上进行过夜培养,挑取单菌落接种至3 mL液体LB培养基中,菌体长出后离心取菌体提取质粒,并将质粒测序验证,将序列正确的质粒转化至大肠杆菌BL21 (DE3) 感受态中,进行后续表达纯化工作。
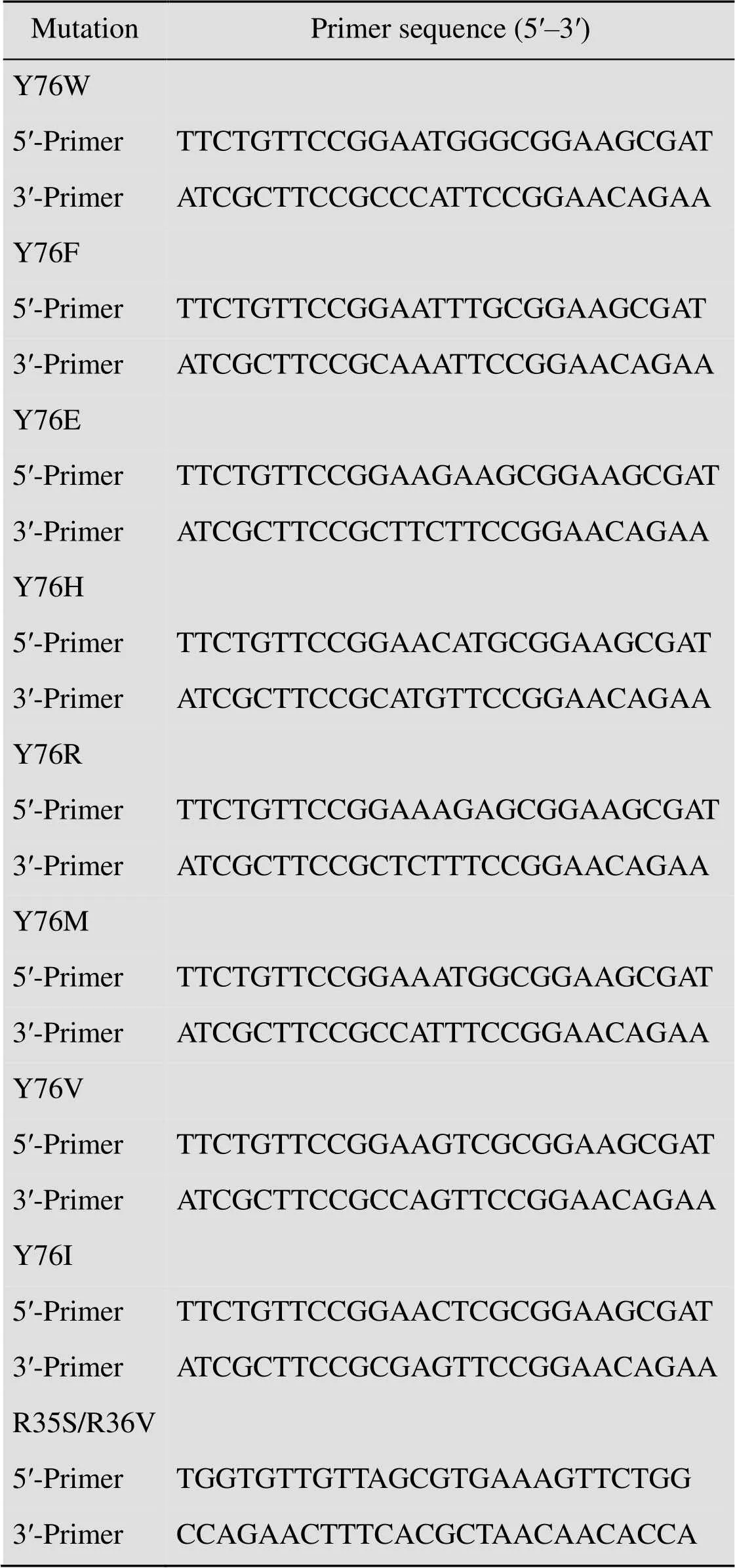
表2 突变引物的序列
1.2.2 突变酶的表达纯化
突变酶的表达、纯化方法和野生型酶类 似[14],首先将含过夜培养的种子液以1%的比例接种至2 000 mL含100 μg/mL氨苄青霉素的液体LB培养基中,于37 ℃、200 r/min培养至600约1.0后,加入诱导剂异丙基-β-D-硫代吡喃半乳糖苷 (IPTG) 至终浓度为0.5 mmol/L,25 ℃、200 r/min继续诱导12 h,4 ℃、5 000 r/min离心30 min收集菌体,菌体用缓冲液A (20 mmol/L Tris-HCl,pH 8.0,500 mmol/L NaCl,5%甘油) 重悬,高压匀浆破碎,破碎液于4 ℃、14 000 r/min离心30 min,收集上清液作为粗酶液,经过 0.22 μm滤膜过滤后进行镍柱亲和层析。镍柱按照说明书进行再生处理并用缓冲液A进行平衡,上样后先用缓冲液A将280紫外吸收洗至基线,再用15%比例的缓冲液B (20 mmol/L Tris-HCl,pH 8.0,500 mmol/L NaCl,5%甘油,500 mmol/L咪唑) 洗脱杂蛋白,接着使用50%比例的缓冲液B洗脱目的蛋白,收集合并洗脱峰,使用Amicon Ultra-15 超滤管 (截留分子量:10 kDa) 进行超滤浓缩,置换缓冲液至缓冲液C (20 mmol/L Tris-HCl,pH 8.0,50 mmol/L NaCl) 并进一步浓缩,浓缩后的酶用于SDS-PAGE以及酶学性质检测。蛋白质浓度使用BCA试剂盒检测,使用牛血清白蛋白BSA作为标准蛋白。
1.2.3 活力测定和动力学参数测定
酶活测定方法参见文献[14],以StDAPDH的天然底物内消旋-二氨基庚二酸 (-DAP) 为底物,以NAD(P)+为辅酶,通过检测340下辅酶NAD(P)H生成时吸光值的变化来表征突变体的活性,辅酶的摩尔吸光系数为 6.22 mmol/(L·cm)。酶活测定体系为:10 mmol/L-DAP,0.5 mmol/L NAD(P)+,200 mmol/L Na2CO3/NaHCO3,pH 10.0,总体积为200 μL。酶活单位 (U) 定义为30 ℃下每分钟生成 1 μmol NAD(P)H所需要的酶量。
对辅酶动力学参数的测定,具体条件如下:100 mmol/L Na2CO3-NaHCO3缓冲液 (pH 10),10 mmol/L-DAP,NADP+终浓度在0.031−16 mmol/L 之间变动,NAD+终浓度在0.031−32 mmol/L之间变动。每个NADP+浓度做3−4次平行反应取平均值,利用GraphPad Prism软件,根据Michaelis-Menten方程式计算野生型酶和各突变体酶对NADP+以及NAD+的表观m和max值。
2 结果与分析
2.1 突变位点的选择及突变设计
从StDAPDH结合辅酶的结构[16]可知,和磷酸基团有直接作用的残基为R35和R36,Y76的位置类似于铁氧化还原蛋白NADP+还原酶中靠近Y258的位置[31-32],StDAPDH结构中这3个位置的氨基酸残基和辅酶的位置关系见图2。与NADP+的腺嘌呤环相临近的76位酪氨酸,其空间位阻较大,且有极性的羟基;为了探索其对辅酶专一性是否有影响,以及是空间位阻还是极性对辅酶专一性产生影响,进行了突变改造,将其突变为和其有类似大位阻的色氨酸 (W)、苯丙氨酸 (F) 和略小位阻的疏水性氨基酸缬氨酸 (V)、甲硫氨酸 (M) 以及异亮氨酸 (I) 以探索空间位阻的影响,突变为亲水氨基酸如碱性的精氨酸 (R)、酸性的谷氨酸 (E) 及组氨酸 (H) 以探索极性的影响。Lerchner等改造醇脱氢酶RasADH的辅酶偏好性从NADP+至NAD+的工作中[25],起始酶与辅酶NADP+上磷酸基团相互作用的氨基酸残基也是两个精氨酸 (R38及R39),通过将它们突变为R38V/R39S后成功将辅酶偏好性改为NAD+依赖型。通过结构比较可以发现,StDAPDH中的R35及R36分别对应RasADH中的R39和R38。对StDAPDH设计了R35S/R36V双突变,探索该双突变是否和RasADH中一样能使辅酶偏好性发生翻转。突变引物序列见表2。
2.2 突变体的纯化以及性质分析
我们首先对构建的Y76单突变进行了表达纯化,利用纯酶对两种辅酶的活力进行了测定及比较,单突变的纯酶结果见图3,其与野生型对两种辅酶的比活力比较见图4。

图2 StDAPDH中和辅酶偏好性可能相关的位点
通过对Y76位的不同单突变对两种辅酶比活力的比较 (图4),我们可以看出,除Y76W外的所有突变子对NAD+的比活力均有所提高,而且所有Y76位突变子对NADP+的比活力均有所下降。在这些突变子中,位阻最大的Y76W对两种辅酶的比活力为Y76位单突变中最低的,可能是因为其较大的位阻影响了NAD(P)+上腺嘌呤的结合,因而影响它们的活力。突变后位阻比酪氨酸略小的疏水氨基酸Y76F对辅酶活力变化趋势和野生型酶影响趋势最为接近,其对NAD+的比活力相比较野生型酶有所提高,其对NADP+的比活力是所有突变子中最高的;酸性氨基酸Y76E对NAD+的活力比Y76F有进一步提高,碱性氨基酸Y76H比Y76E对NAD+的活力更高,但对NADP+的活力略低;同为碱性的氨基酸Y76R比Y76H对NAD+的活力则更高一些,对NADP+的活力也相对更高;位阻更小的疏水性的Y76M、Y76V以及Y76I对NAD+的活力逐步增高,对NADP+的活力也相应逐步降低,表明它们的辅酶偏好性偏转能力也逐步提高;在我们测试的这些Y76突变子中,Y76V及Y76I对NAD+的比活力最高。在所有Y76的突变子中,虽然Y76E有最高的NAD+/NADP+比值 (0.73),但其对NAD+的活力并不是最高;而Y76I突变子,虽然NAD+/NADP+比值为0.69,但其对NAD+活力是Y76突变子中最高的。
为考察R35/R36为对辅酶偏好性的影响,我们借鉴别人的结果,进行了R35S/R36V双突变;为考察Y76位的突变和R35、R36位突变是否有相互作用,我们选取了Y76单突变中对NADP+活力最高的Y76F,对NAD+活力最高的Y76V和Y76I,分别与R35S/R36V进行了组合突变,并进行了表达、纯化及活力测定。纯化结果见图5。

图3 StDAPDH Y76单突变纯酶的SDS-PAGE结果

图4 野生型和Y76单突变酶的比活力
R35S/R36V双突变对StDAPDH辅酶偏好性影响和Y76W单突变类似,对两个辅酶的活力都有明显降低,而且对NAD+的比活力仍然低于对NADP+的比活力 (图6)。对R35、R36两个位点的突变结果表明,尝试改造的R35S/R36V虽然确实对酶的偏好性有影响,其NAD+/NADP+比值 (1.25) 比所有Y76单突变 (最高值为0.73) 都高,但是对NAD+活力提高不明显。R35S/R36V双突变和Y76F组合后的R35S/R36V/Y76F对辅酶NAD+的比活力相比 Y76F单突变没有明显变化,但NAD+/NADP+比值却从0.39提高到1.25,而且该突变对NAD+的比活力高于对NADP+的比活力;R35S/R36V和Y76I及Y76V组合后的突变子对NAD+的比活力也均高于它们对NADP+的比活力,而且它们的NAD+/NADP+比值也均有明显提高,分别从0.69到1.39,以及0.53到1.36;在单突变中Y76I比Y76V对NAD+的比活力稍高,但在组合的三突变中,R35S/R36V/Y76V却比R35S/R36V/Y76I对NAD+的比活力更高。这些结果表明,R35/R36位和Y76位组合后,可以获得比单独进行单突变或双突变更好的结果,但其所起的作用并不是简单的叠加。后续若想获得更好结果,可能需要先筛选出R35/R36位较好的结果,并将较好的双突变结果和76位进行进一步组合突变并进行筛选。

图5 R35S/R36V以及三突变纯酶的SDS-PAGE 结果

图6 野生型和突变体酶的比活力
本研究中涉及的突变子动力学参数测定结果见表3。从表中可以看出,所有Y76突变对NAD+的m都降低了,而且除了Y76E外所有突变对NADP+的m也降低了,其中对NADP+的m值最小的是Y76F,这说明Y76F对NADP+的亲和力最好;Y76E对NADP+的cat最高,但同时其m也最高,这说明Y76E在提高了其催化效率的同时也降低了对NADP+的亲和力;对NAD+的m值最小的是空间位阻较小的Y76M、Y76I和Y76V,Y76V对NAD+的cat和cat/m是所有突变中最大的,这说明在对NAD+的亲和力和催化效率中,空间位阻起到了重要的作用。
从表3中还可以看出,R35S/R36V对NADP+和NAD+的m、cat以及cat/m都降低了,这说明与磷酸基团有相互作用的R35、R36对辅酶亲和力和催化效率有非常重要的作用。同时,还可以看出Y76对R35S/R36V的作用与Y76的单突变的效果一致,当Y76的空间位阻依次变小 (F、I、V) 时,其对NAD+的m也变小;通过比较单突变Y76V,Y76I和三突变R35S/R36V/Y76V和R35S/R36V/Y76I对NAD+的cat/m时可以发现,它们之间存在协同作用:Y76V的cat/m比Y76I大,但R35S/R36V/Y76V的cat/m却比R35S/R36V/Y76I的数值小,这可能是因为Y76V和R35S/R36V都突变为位阻较小的氨基酸残基,表3中m(NAD+)/m(NADP+) 值显示,Y76突变对NAD+和NADP+亲和力差异的贡献各不相同,与野生型相比,空间位阻相对较小的M、V和I对辅酶的亲和力更偏好于NAD+,其中Y76I的m(NAD+)/m(NADP+) 值比野生型的低了6倍多,显示其对NAD+的亲和力有明显的提高。双突变R35S/R36V与野生酶相比,m(NAD+)/m(NADP+) 值相差23倍,在此基础上,增加额外的一个突变 (无论是Y76I,F或者V) 都导致此效果的进一步提升。表3中cat(NAD+)/cat(NADP+) 的结果显示,与野生酶相比,大部分突变都导致了酶对NAD+和NADP+的cat差异的变化,其中以Y76R和Y76V导致酶选择NAD+作为辅因子最有效,3个突变中,R35S/R36V/Y76I最高。
以上结果分析可知,R35、R36对NAD+和NADP+偏好性的选择具有关键的作用,这可能是其直接与磷酸基团相互作用的结果。不仅如此,Y76对NAD+和NADP+的偏好性选择也有重要的作用,可能通过对嘌呤环的作用,改变了NAD+在酶结构中的构象,使NAD+更好地与酶结合,导致催化效率的提高。

表3 野生型StDAPDH和所有突变子的动力学参数
3 讨论
从上述结果中可以发现,虽然Y76单突变对NAD+的比活仍低于对NADP+的比活,但该位点的突变对辅酶偏好性确有影响,动力学数据结果表明,Y76位的突变是通过改变酶和辅酶NADP+和NAD+的亲和力来影响其辅酶专一性的。对与NADP+磷酸基团有直接作用的R35/R36设计的R35S/R36V突变,虽然改变了辅酶偏好性,但对NAD+的比活力没有明显提升;而测试的Y76突变和R35S/R36V突变组合,对辅酶偏好性有影响的同时,对NAD+的比活力均已大于对NADP+的比活力。这些结果表明,距离腺嘌呤较近的Y76对StDAPDH的辅酶偏好性确有影响,而且该位置和与NADP+上磷酸有直接作用的R35及R36对辅酶偏好性有协同作用。
在本文进行的改造尝试基础上,对StDAPDH的R35、R36及Y76进行组合饱和突变,也许能获得对NAD+活力更高的突变酶,相关研究工作正在进行中。
[1] Yonaha K, Soda K. Applications of stereoselectivity of enzymes: synthesis of optically active amino acids and alpha-hydroxy acids, and stereospecific isotope labeling of amino acids, amines and coenzymes. Adv Biochem Eng Biotechnol, 1986, 33: 95−130.
[2] Tanigawa M, Shinohara T, Saito M, et al. D-Amino acid dehydrogenase fromNCTC 11637. Amino Acids, 2010, 38(1): 247−255.
[3] Xu SJ, Ju JS, Ma YH. Identification and expression a D-amino acid dehydrogenase gene fromTM5-2. Acta Microbiol Sin, 2007, 47(4): 634−638 (in Chinese).徐书景, 鞠建松, 马延和.荧光假单胞菌TM5-2中D-型氨基酸脱氢酶的鉴定和表达. 微生物学报, 2007, 47(4): 634−638.
[4] Jones H, Venables WA. Solubilisation of D-amino acid dehydrogenase ofK12 and its re-binding to envelope preparations. Biochimie, 1983, 65(3): 177−183.
[5] Jones H, Venables WA. Effects of solubilisation on some properties of the membrane-bound respiratory enzyme D-amino acid dehydrogenase ofFEBS Lett, 1983, 151(2): 189−192.
[6] Wild J, Obrepalska B. Regulation of expression of theA gene encoding D-amino acid dehydrogenase in: analysis ofA-fusions and direction of dadA transcription. Mol Gen Genet, 1982, 186(3): 405−410.
[7] Wild J, Klopotowski T. D-amino acid dehydrogenase ofK12: positive selection of mutants defective in enzyme activity and localization of the structural gene. Mol Gen Genet, 1981, 181(3): 373−378.
[8] Olsiewski PJ, Kaczorowski GJ, Walsh C. Purification and properties of D-amino acid dehydrogenase, an inducible membrane-bound iron sulfur flavoenzyme fromB. J Biol Chem, 1980, 255(10): 4487−4494.
[9] Misono H, Soda K. Properties of-alpha, epsilon-diaminopimelate D-dehydrogenase from. J Biol Chem, 1980, 255(22): 10599−10605.
[10] Vedha-Peters K, Gunawardana M, Rozzell JD, et al. Creation of a broad-range and highly stereoselective D-amino acid dehydrogenase for the one-step synthesis of D-amino acids. J Am Chem Soc, 2006, 128(33): 10923−10929.
[11] Akita H, Doi K, Kawarabayasi Y, et al. Creation of a thermostable NADP(+)-dependent D-amino acid dehydrogenase fromstrain A1-diaminopimelate dehydrogenase by site-directed mutagenesis. Biotechnol Lett, 2012, 34(9): 1693−1699.
[12] Akita H, Suzuki H, Doi K, et al. Efficient synthesis of D-branched-chain amino acids and their labeled compounds with stable isotopes using D-amino acid dehydrogenase. Appl Microbiol Biotechnol, 2014, 98(3): 1135−1143.
[13] Misono H, Togawa H, Yamamoto T, et al.-alpha, epsilon-diaminopimelate D-dehydrogenase: distribution and the reaction product. J Bacteriol, 1979, 137(1): 22−27.
[14] Gao X, Chen X, Liu W, et al. A novel-diaminopimelate dehydrogenase from: overexpression, characterization, and potential for D-amino acid synthesis. Appl Environ Microbiol, 2012, 78(24): 8595−8600.
[15] Gao X, Huang F, Feng J, et al. Engineering the-diaminopimelate dehydrogenase fromby site-saturation mutagenesis for D-phenylalanine synthesis. Appl Environ Microbiol, 2013, 79(16): 5078−5081.
[16] Liu W, Li Z, Huang CH, et al. Structural and mutational studies on the unusual substrate specificity of-diaminopimelate dehydrogenase from. Chem Biochem, 2014, 15(2): 217−222.
[17] Weckbecker A, Groger H, Hummel W. Regeneration of nicotinamide coenzymes: principles and applications for the synthesis of chiral compounds. Adv Biochem Engin/Biotechnol, 2010, 120: 195−242.
[18] Banta S, Swanson BA, Wu S, et al. Alteration of the specificity of the cofactor-binding pocket of Corynebacterium 2,5-diketo-D-gluconic acid reductase A. Protein Eng, 2002, 15(2): 131−140.
[19] Liang L, Zhang J, Lin Z. Altering coenzyme specificity ofxylose reductase by the semi-rational approach CASTing. Microb Cell Fact, 2007, 6: 36.
[20] Rosell A, Valencia E, Ochoa WF, et al. Complete reversal of coenzyme specificity by concerted mutation of three consecutive residues in alcohol dehydrogenase. J Biol Chem, 2003, 278(42): 40573−40580.
[21] Khoury GA, Fazelinia H, Chin JW, et al. Computational design ofxylose reductase for altered cofactor specificity. Protein Sci, 2009, 18(10): 2125−2138.
[22] Cui DB, Zhang LJ, Yao ZQ, et al. Computational design of short-chain dehydrogenase Gox2181 for altered coenzyme specificity. J Biotecnol, 2013, 167(4): 386−392.
[23] Scrutton NS, Berry A, Perham RN. Redesign of the coenzyme specificity of a dehydrogenase by protein engineering. Nature, 1990, 343(6253): 38−43.
[24] Cho H, Oliveira MA, Tai HH. Critical residues for the coenzyme specificity of NAD(+)-dependent 15-hydroxyprostaglandin dehydrogenase. Arch Biochem Biophys, 2003, 419(2): 139−146.
[25] Lerchner A, Jarasch A, Meining W, et al. Crystallographic analysis and structure-guided engineering of NADPH-dependentsp alcohol dehydrogenase toward NADH cosubstrate specificity. Biotechnol Bioeng, 2013, 110(11): 2803−2814.
[26] Rane MJ, Calvo KC. Reversal of the nucleotide specificity of ketol acid reductoisomerase by site-directed mutagenesis identifies the NADPH binding site. Arch Biochem Biophys, 338(1): 83–89.
[27] Zeng QK, Du HL, Wang JF, et al. Reversal of coenzyme specificity and improvement of catalytic efficiency ofxylose reductase by rational site-directed mutagenesis. Biotechnol Lett, 2009, 31(7): 1025−1029.
[28] Bubner P, Klimacek M, Nidetzky B. Structure guided engineering of the coenzyme specificity ofmannitol 2-dehydrogenase to enable efficient utilization of NAD(H) and NADP(H). FEBS Lett, 2008, 582(2): 233−237.
[29] Dambe TR, Kuhn AM, Brossette T, et al. Crystal structure of NADP(H)-dependent 1,5-anhydro-D-fructose reductase fromat 2.2 Å resolution: construction of a NADH-accepting mutant and its application in rare sugar synthesis. Biochemistry, 2006, 45(33): 10030−10042.
[30] Shiraishi N, Croy C, Kaur J, Campbell WH. Engineering of pyridine nucleotide specificity of nitrate reductase: mutagenesis of recombinant cytochrome b reductase fragment ofNADPH: nitrate reductase. Arch Biochem Biophys,1998, 358(1): 104–115.
[31] Crobu D, Canevari G, Milani M, et al.ferredoxin-NADP+reductase His286 plays a dual role in NADP(H) binding and catalysis. Biochemistry, 2009, 48(40): 9525−9533.
[32] Baroni S, Pandini V, Vanoni MA, et al. A single tyrosine hydroxyl group almost entirely controls the NADPH specificity offerredoxin-NADP+reductase. Biochemistry, 2012, 51(18): 3819−3826.
(本文责编 陈宏宇)
Effect of residue Y76 on co-enzyme specificity of-diaminopimelate dehydrogenase from
Leiming Zhao1,2, Weidong Liu2, Xi Chen2, Min Wang1, Jinhui Feng2, Qiaqing Wu2, and Dunming Zhu2
1 Key Laboratory of Industrial Microbiology Fermentation of Ministry of Education, College of Biotechnology, Tianjin University of Science & Technology, Tianjin 300457, China 2 Tianjin Biocatalysis Technology Engineering Center, National Engineering Laboratory for Industrial Enzymes, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, China
Inindustrial application of NAD(P)H-dependent dehydrogenases, NAD(H) has the advantages over NADP(H) in higher stability, lower price and wider recycling system. Recently, ao-2,6-diaminopimelate dehydrogenase from(StDAPDH) has been found to be a useful biocatalyst for the production of D-amino acids, but it requires NADP(H) as co-enzyme. To switch the co-enzyme specificity from NADP(H) to NAD(H), we studied the effect of Y76 on the co-enzyme specificity of StDAPDH, because the crystal structural analysis indicated that residue Y76 is near the adenine ring. The mutation of Y76 exerted significant effect on the co-enzyme specificity. Furthermore, the double mutant R35S/R36V significantly lowered the specific activity toward NADP+, and the combination of R35S/R36V with some of the Y76 mutants resulted in mutant enzymes favorable NAD+over NADP+. This study should provide useful guidance for the further development of highly active NAD+-dependent StDAPDH by enzyme engineering.
amino acid dehydrogenase,-diaminopimelate dehydrogenase, co-enzyme preference, mutagenesis
October 30, 2014;
January 19, 2015
Key Deployment Project of Chinese Academy of Sciences (No. KSZD-EW-Z-016-3), National Natural Science Foundation of China (Nos. 21072151, 21102100), National Basic Research Program of China (973 Program) (No. 2011CB710801).
:Qiaqing Wu. Tel: +86-22-84861963; Fax: +86-22-84861996; E-mail: wu_qq@tib.cas.cn Dunming Zhu. Tel: +86-22-84861962; Fax: +86-22-84861996; E-mail: zhu_dm@tib.cas.cn
中国科学院重点部署项目 (No. KSZD-EW-Z-016-3),国家自然科学基金 (Nos. 21072151, 21102100),国家重点基础研究发展计划 (973计划) (No. 2011CB710801) 资助。
