新型亲和去垢小柱净化-液相色谱-串联质谱法分析水稻叶片蛋白质组
2014-12-26曹晓林巩佳第陈铭学于莎莎卞英芳曹赵云
曹晓林, 巩佳第, 陈铭学, 于莎莎, 卞英芳, 曹赵云
(中国水稻研究所,农业部稻米及制品质量监督检验测试中心,农业部稻米产品质量安全风险评估实验室,浙江 杭州310006)
鸟枪法蛋白质组学是继传统的凝胶电泳-质谱技术之后的另一种新型蛋白质分析策略。近年来,随着色谱-质谱技术和生物信息学的迅速发展[1-3],以及各种蛋白质数据库的日益完善,鸟枪法蛋白质组学成为分析复杂蛋白质组的有力工具[4,5],并已成功地用于水稻等重要作物的器官发育调控机理、逆境胁迫生理和农产品质量安全等热点研究领域[6,7]。然而,鸟枪法蛋白质组学中,蛋白质样品制备方法仍存在亟待解决的问题,突出表现在蛋白质离液剂选择和对其去除的矛盾上。
蛋白质组的准确鉴定高度依赖于蛋白质的裂解、变性程度等[8]。虽然十二烷基硫酸钠(sodium dodecyl sulfate,SDS)裂解法是当前使用最为广泛和有效的蛋白质(尤其是疏水膜蛋白质)裂解技术[9,10],但蛋白质裂解液中残留的SDS 会显著降低酶解效率、干扰肽段的色谱分离并对目标物离子化过程产生强烈的离子抑制作用[11,12]。由此可见,研究建立高效的SDS 净化方法是解决上述矛盾的关键。目前针对SDS 的净化主要依赖于传统的丙酮沉淀法[13]、超滤辅助样品制备(filter aided sample preparation,FASP)法[9]以及各种色谱法等。尽管这些技术均有较好的去除SDS 效果,但通常会引起疏水性蛋白质的损失[9,14],且存在步骤繁琐、操作耗时、分析通量低等问题。亲和去垢小柱是近年来推出的一种新型净化小柱,利用内部树脂填料的疏水性空腔可与离液剂形成配合物的特性达到将其去除的目的[15]。该小柱由于对SDS 吸附作用强,引起目标蛋白质损失小等优点已日益引起人们的关注,然而,目前仅在针对动物和微生物蛋白质处理中有少量报道[16],尚未见其在植物样品中的应用。
本实验将该新型亲和去垢小柱用于水稻蛋白质样品的净化。考察了传统的FASP 法、丙酮沉淀法和新型亲和去垢小柱法对SDS 的去除效果,比较了上述3 种净化技术对水稻叶片蛋白质鉴定结果(蛋白质数目、相对分子质量和等电点分布)的影响,最终建立了以亲和去垢小柱净化,纳升液相色谱-线性离子阱/静电场轨道阱质谱联用的水稻叶片蛋白质分析方法,以期为水稻蛋白质样品的净化及开展相关蛋白质组学研究提供重要技术参考。
1 实验部分
1.1 仪器与设备
Easy nLC 1000 纳升级液相色谱仪、LTQ-Orbitrap 组合高分辨质谱仪(附纳喷雾离子源)、Biofuge Primo R 型台式冷冻离心机(美国ThermoFisher 公司)。冷 冻 干 燥 机 FreeZone 2.5 Plus (美 国LABCONCO 公司);分光光度计(上海光谱仪器有限公司);PHS-3C 精密pH 计(上海仪电科学仪器股份有限公司);电热恒温振荡水槽DK2-28(上海精宏实验设备有限公司)。
1.2 试剂与材料
Tris-HCl、SDS、碘 代 乙酰 胺(iodoacetamide,IAA)、PBS 缓冲液(phosphate buffer saline)、胰蛋白酶(trypsin)和蛋白酶抑制剂均购于Sigma 公司;碳酸氢铵购于Acros Organics 公司;二硫苏糖醇(dithiothreitol,DTT)购于Promega 公司;乙腈、甲醇购于德国Merck 公司;Tris 饱和酚、SDS 残留检测试剂盒购于Sangon 公司;以上试剂均为分析纯或色谱纯。实验室用水为Milli-Q 高纯水(电阻率为18.2 MΩ·cm);亲和去垢小柱(pierce affinity detergent removal spin columns)购自Thermo 公司;C18-SD 除盐小柱购自Agilent 公司。水稻品种镇稻11 号由中国水稻研究提供,在中国水稻研究所网室进行常规土培,采集完熟期水稻叶片,用液氮速冻并在-80 ℃冰箱中保存,待用。
1.3 样品的制备
1.3.1 叶片总蛋白质的提取
叶片总蛋白质提取参考Sheoran 等[17]报道的酚提取法进行。称取3 g 经液氮研磨后的水稻叶片样品,加入20 mL 提取缓冲液(含100 mmol/L KCl、0.7 mol/L 蔗 糖、50 mmol/L Tris-HCl、5 mmol/L EDTA、10 μL 蛋白酶抑制剂),振荡混匀,加入1 mol/L DTT 200 μL,于4 ℃振荡30 min;加入等体积的Tris 饱和酚,混匀后于4 ℃、6 000 g 条件下离心30 min;取上层酚相,加入等体积提取缓冲液,重复上述离心步骤一次;最后取出上层酚相,加入3 倍体积的预冷甲醇溶液(含0.1 mol/L 乙酸铵),于-20℃静置过夜;再按上述条件离心,弃上清液;分别用甲醇和丙酮清洗沉淀两次,冷冻干燥。
1.3.2 蛋白质的裂解
参照Sheoran[17]方法,略有改进。取上述蛋白质样品1 mg,加入450 μL 40 g/L SDS 溶液(含100 mmol/L Tris-HCl,pH 8.0),95 ℃中加热3 min,再加入1 mol/L DTT 至终浓度为0.1 mol/L,置于37℃水浴中加热2.5 h,取出,待净化。
1.3.3 蛋白质的净化及酶解
1.3.3.1 亲和去垢小柱法
取100 μL蛋白质裂解溶液,加100 μL水混匀。将亲和去垢小柱置于2 mL 离心管中,于1 500 g 离心1 min,以除去原有的储存溶液;向亲和去垢小柱内加入400 μL平衡缓冲溶液(PBS 溶液),于1 500 g 离心1 min,弃流出液,重复一次;将上述稀释后的裂解液移入亲和去垢小柱内,常温放置2 min 后,于1 500 g 离心2 min,收集流出液;取出少许,待测SDS 残留量;其余部分用10 mmol/L NH4HCO3稀释至500 μL,并加入胰蛋白酶4 μg,于37 ℃水浴酶解16 h;酶解液分别经C18-SD 脱盐和冷冻干燥,最后用100 μL 0.1%(v/v)甲酸溶液溶解,待上机。
1.3.3.2 FASP 法
参考Wis'niewski 等[18]和Manza 等[19]的方法:取裂解还原后的蛋白质溶液100 μL,用8 mol/L 的尿素(以100 mmol/L Tris-HCl 配制,pH 8.5)稀释至SDS 质量浓度为1 g/L,备用;用10 kDa 超滤离心管超滤离心(4 ℃、6 000 g)30 min 经稀释后的蛋白质溶液,每次约4 mL;再加入2 mL 8 mol/L 尿素(以100 mmol/L Tris-HCl 配制,pH 8.0),相同条件下超滤离心,重复3 次;合并流出液,测SDS 残留量;最后将尿素置换后酶解,后续操作同1.3.3.1 节。
1.3.3.3 丙酮沉淀法
参考Lin 等[13]方法:取裂解还原后的蛋白质溶液100 μL,加入600 μL预冷丙酮,涡旋振荡,置于-20℃冰箱中过夜;于4 ℃、10 000 g 条件下离心40 min;取出上清液,将离心管开口倒置于微尘纸上,至残余液体流出;再加入1 mL 冷丙酮,涡旋振荡1 min,重复上述步骤;合并两次上清液用于测定SDS 的残留量;样品经低温干燥、酶解过夜。将酶解后的溶液加入到10 kDa 超滤管中,于室温、6 000 g 离心30 min;再加入200 μL 50 mmol/L 碳酸氢铵,于室温、6 000 g 离心30 min;后续操作同1.3.3.1 节。
1.3.4 SDS 的残留定量
对3 种方法净化后酶解前的流出液进行SDS的残留定量,参照SDS 残留试剂盒说明书进行。
1.4 仪器条件
色谱分析柱为Acclaim Pep Map RSLC(C18,15 cm×50 μm,2 μm);预柱为Acclaim Pep Map 100(C18,2 cm×75 μm,3 μm)。柱温30 ℃;流动相A为0.1%(v/v)甲酸,B 为乙腈。梯度洗脱程序:0 ~120 min,0% B ~65% B;120 ~140 min,65% B ~95%B;140 ~160 min,95% B。流速为300 nL/min,进样量为2 μL。
设置电喷雾电压为2 kV,离子传输毛细管的温度为200 ℃。以数据依赖模式(data-dependent acquisition mode)进行MS/MS 图谱采集。一级质谱采用Orbitrap 采集,扫描范围为m/z 400 ~1 800,分辨率设为60 000;对全扫描中丰度最高的10 个离子峰用LTQ 进行二级碎片采集,裂解模式为CID(collision induced dissociation),归一化碰撞能量(normalized collision energy)为35%,离子活化时间(activation time)为30 ms。动态排除(dynamic exclusion)设置:重复次数(repeat count)为2,重复间隔时间(repeat duration)为30 s,动态排除时间(exclusion duration)为90 s。
1.5 数据采集及处理
采用Xcalibur 软件(Version 2.07,Thermo 公司)进行系统控制和数据收集。质谱采集数据在水稻蛋白质数据库(Oryza_sativa Uniprot. fasta)中进行检索,数 据库 来 源 于http://www. ncbi. nlm. nih.gov/protein/term =Oryza +sativa;数据库检索软件为SEQUEST;所有肽段设置胰蛋白酶全酶切,最高允许两个漏切位点;设定半胱氨酸残基C 端为固定修饰+57 Da,Met(M)为动态修饰+15.995 Da;前体离子的质量容忍度设置为10 ppm,碎片离子的质量容忍度设置为1 Da。假阳性率控制在1% 以内,以此获取更为可靠的鉴定结果。采用Excel 软件进行数据分析。
2 结果与讨论
2.1 不同净化方法的SDS 去除效率及对蛋白质鉴定结果的影响
水稻叶片蛋白质经提取、裂解后,分别按FASP法、亲和去垢小柱法和丙酮沉淀法进行净化及随后的蛋白质鉴定,实验分别考察了其SDS 去除效率以及对蛋白质鉴定结果的影响。结果表明,FASP 法、亲和去垢小柱法和丙酮沉淀法均有较好的SDS 去除效果,各自对应的SDS 去除效率分别达98.7%、95.9%和97.3% (n =3)。三者鉴定得到蛋白质数目为196、563 和306 种,其中亲和去垢小柱法鉴定到的蛋白质数目最多,分别是FASP 法和丙酮沉淀法的2.8 和1.8 倍。说明FASP 法、丙酮沉淀法在裂解液净化过程中均造成明显的蛋白质损失,最终导致各自检测到的蛋白质数目减少。其主要原因可能是前者所采用的超滤膜对一些蛋白质(尤其是膜蛋白质)具有较强的吸附作用[20,21],而丙酮沉淀法的损失主要与蛋白质沉淀不完全有关[22]。对于亲和去垢小柱法而言,除鉴定蛋白质数目多以外,还具有过程简单、操作步骤少和高通量等技术优势。
任取各自方法的一次蛋白质分析结果,研究了鉴定蛋白质种类上的互补性(见图1)。3 种方法共鉴定出807 种非冗余蛋白质,其中亲和去垢小柱法可鉴定出565 种,占总鉴定蛋白质种类的70% 以上;而FASP 法和丙酮沉淀法则分别鉴定出92 和124 种蛋白质,仅约占鉴定总数的11.4%和15.4%,另外的26 种由它们共同鉴定得到,约占总数的3.2%。可见,尽管3 种方法鉴定的蛋白质存在一定的互补性,但亲和去垢小柱法在鉴定蛋白质种类上占绝对优势。
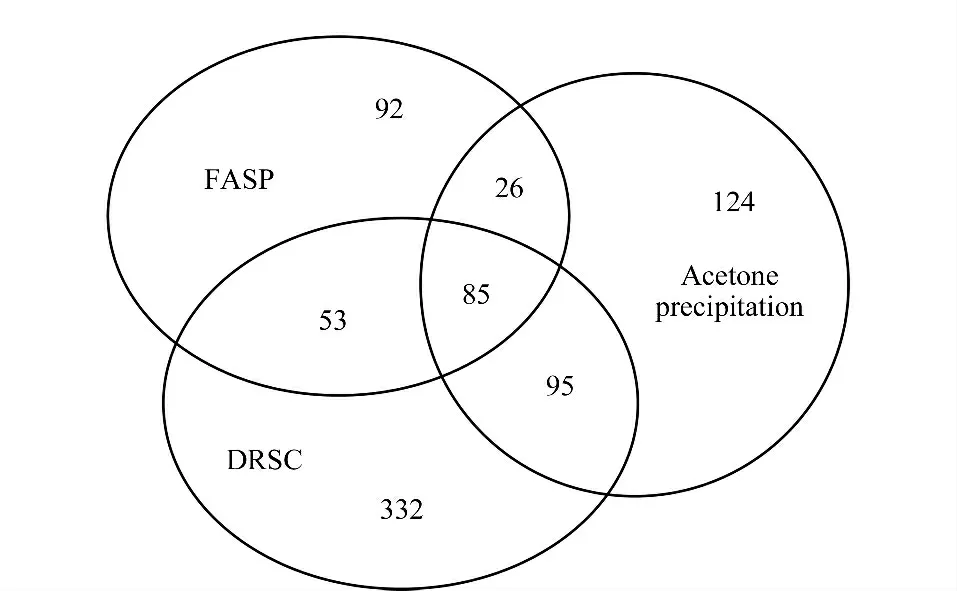
图1 FASP 法、亲和去垢小柱法和丙酮沉淀法在鉴定蛋白质种类上的互补性Fig.1 Complementary analysis of identified proteins by filter aided sample preparation (FASP),detergent removal spin column (DRSC)and acetone precipitation methods
2.2 不同净化方法所鉴定的蛋白质相对分子质量及等电点的分布
在蛋白质组学研究中,某个特定等电点(pI)或相对分子质量(MW)范围的蛋白质通常更受关注[13]。为此,本文对FASP 法、亲和去垢小柱法和丙酮沉淀法各自鉴定到的蛋白质按MW和pI 进行分析比较,以反映这3 种净化方法对不同MW和pI蛋白质的偏好性。
图2 比较了3 种净化方法所鉴定到蛋白质在不同pI 值区间的数目。可以看出,亲和去垢小柱法在不同pI 区间所鉴定到的蛋白质均多于FASP 法和丙酮沉淀法,但3 种方法所鉴定的蛋白质占各自鉴定蛋白质数目的比例却基本相同。在pI 6 ~9 之间,3种方法对应的蛋白质数目分别为91、257、135,占各自鉴定蛋白质总数目的45% 左右;类似地,pI 3 ~6和pI≥9 时,3 种方法鉴定的数目均分别占各自鉴定总数目的35% 和18% 左右。由此说明3 种方法对不同pI 值的蛋白质均没有明显的选择性。

图2 3 种净化方法鉴定到的蛋白质在不同等电点区间的分布Fig.2 Distribution of the identified proteins in different intervals of pI by three different purification methods
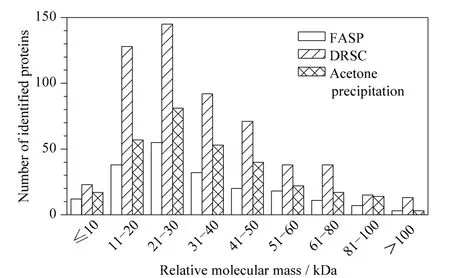
图3 3 种净化方法鉴定到的蛋白质在不同相对分子质量区间的分布Fig.3 Distribution of the identified proteins in different intervals of relative molecular masses by three different purification methods
图3 表明了3 种净化方法鉴定到的蛋白质在不同MW区间的分布情况。可以看出,其结果与上述pI 值分布类似,即3 种净化方法所鉴定蛋白质占各自蛋白质鉴定总数的比例总体上没有明显差异,且鉴定到的水稻叶片蛋白质的相对分子质量均主要集中在11 ~50 kDa,与Muthurajan 等[23]和Zhao 等[24]的研究结果基本一致。由此可见,在针对不同相对分子质量和pI 值蛋白质的净化中,采用亲和去垢小柱法均能取得满意的结果,而FASP 法和丙酮沉淀法对不同相对分子质量和pI 值蛋白质造成类似程度的损失。
2.3 水稻叶片蛋白质鉴定结果分析
在最佳的色谱、质谱条件下,水稻叶片蛋白质经提取、SDS 裂解、亲和去垢小柱净化,酶解肽段经纳升级液相色谱-高分辨质谱分析(典型的基峰色谱图见图4),再经相关数据库检索,一次进样分析可鉴定多达588 种蛋白质。
图4 水稻叶片蛋白酶解液的基峰色谱图Fig.4 Base peak chromatogram of protein trypsin degradation from rice leaves
研究表明,鉴定到2 个及以上的完整肽段,则其蛋白质鉴定结果具有极高的可信度[25]。为此,本文按肽段匹配数对563 种叶片蛋白质(n =3)进行分析,结果表明肽段匹配数≥2 的有296 种,占总数的一半以上,最多时肽段匹配数可达16 个。对肽段匹配数≥2 的296 种蛋白质,按照其生物学功能进行了分类(见图5),其中主要包括结合活性(与ATP、镁离子、钙离子、锌离子、铜离子、核糖核苷酸、血红素等结合)、酶活性(异构酶、氧化还原、水解酶、合成酶等)、转移运输活性和结构组成功能等,对应的数量分别为113、90、20 和21 种。
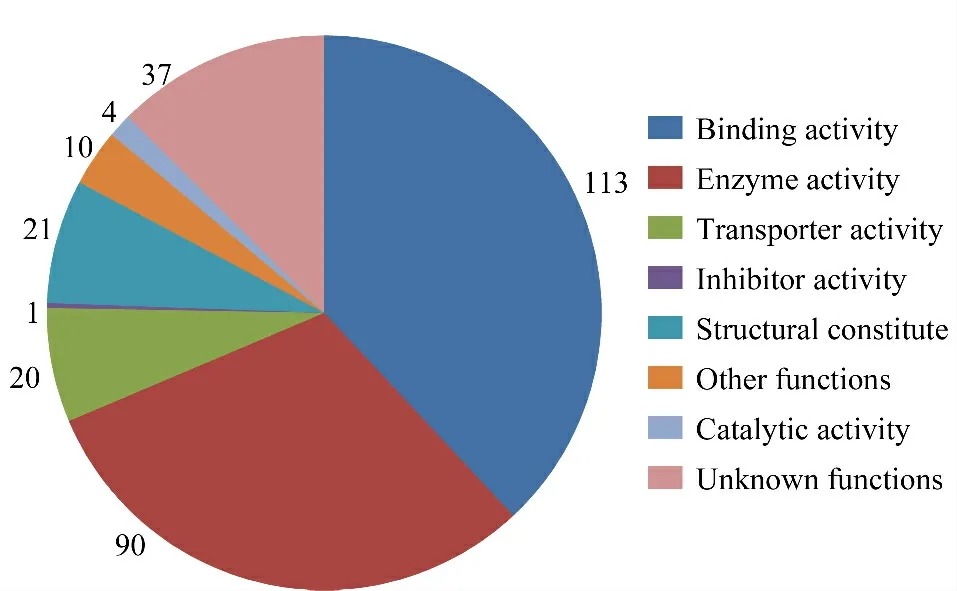
图5 水稻叶片中蛋白质分子功能的分类Fig.5 Distribution of proteins of rice leaves classified by different molecular functions
表1 列举了相对分子质量为9.3 ~177.2 kDa和pI 为4.26 ~11.47 的4 种典型蛋白质,其肽段匹配率(coverage)均在9%以上,且每种蛋白质都含有2 个及以上的完整鉴定肽段(identified peptide numbers),弥补了传统的凝胶电泳对极性较大蛋白质、相对分子质量较小或较大蛋白质不能很好分离鉴定的不足。此外,核酮糖二磷酸羧化酶是评价叶片蛋白质鉴定方法的特征蛋白质[7],序列号(Accession No.)为Q339G9 的核酮糖二磷酸羧化酶大亚基是该酶的重要组成部分;本文中该亚基对应的肽段匹配数为12,肽段匹配率为29.01%,明显优于Islam等[26]肽段匹配数为5 的结果。

表1 4 种典型水稻叶片蛋白质的鉴定结果Table 1 Identification results of four typical proteins in rice leaves
3 结论
本文建立了基于亲和去垢小柱净化的水稻叶片蛋白质分析鉴定方法。通过比较FASP 法、亲和去垢小柱法和丙酮沉淀法对SDS 去除效率及对蛋白质鉴定结果的影响,发现3 种方法均有较好的SDS去除效果(去除效率均>95%);尽管3 种方法具有一定互补性,但以亲和去垢小柱法鉴定的蛋白质总数最多,分别是FASP 法和丙酮沉淀法的2.8 倍和1.8 倍。亲和去垢小柱法极大地简化了蛋白质样品制备的步骤,提高了结果的可靠性,实现了高通量分析,并对不同相对分子质量和pI 值的蛋白质均具有较好的净化效果,一次进样分析可鉴定到588 种水稻叶片蛋白质。本方法不仅可为开展水稻叶片蛋白质组学研究提供重要的技术支撑,还可为其他植物蛋白质组学的研究技术提供重要的借鉴。
[1] Second T P,Blethrow J D,Schwartz J C,et al. Anal Chem,2009,81:7757
[2] Hsieh E J,Hoopmann M R,MacLean B,et al. J Proteome Res,2010,9:1138
[3] Yu H Y,Yan J Z,Guo M,et al. Chinese Journal of Chromatography (于海洋,晏嘉泽,郭明,等. 色谱),2013,31(4):362
[4] Yates J R. J Am Chem Soc,2013,135(5):1629
[5] Jorrin-Novo J V,Komatsu S,Weckwerth W,et al. Methods in Molecular Biology. New York:Humana Press,2014
[6] Li B,Ning L Y,Zhang J W,et al. Hubei Agricultural Sciences(李蓓,宁露云,张俊卫,等. 湖北农业科学),2013,52(32):5403
[7] Kim S T,Kim S G,Agrawal G K,et al. Proteomics,2014,14:593
[8] Masuda T,Tomita M,Ishihama Y. J Proteome Res,2008,7:731
[9] Bereman M S,Egertson J D,MacCoss M J. Proteomics,2011,11:2931
[10] Winter D,Steen H. Proteomics,2011,11:4726
[11] Tanca A,Biosa G,Pagnozzi D,et al. Proteomics,2013,13:2597
[12] Zhang N,Chen R,Young N,et al. Proteomics,2007,7:484[13] Lin Y,Liu H,Liu Z,et al. J Chromatogr B,2012,901:18
[14] Botelho D,Wall M J,Vieira D B,et al. J Proteome Res,2010,9:2863
[15] Antharavally B S,Mallia K A,Rosenblatt M M,et al. Anal Biochem,2011,416:39
[16] Hengel S M,Floyd E,Baker E S,et al. Proteomics,2012,12:3138
[17] Sheoran I S,Ross A R S,Olson D J H,et al. Plant Sci,2009,176:99
[18] Wis'niewski J R,Zougman A,Nagaraj N,et al. Nat Methods,2009,6:359
[19] Manza L L,Stamer S L,Ham A J L,et al. Proteomics,2005,5:1742
[20] Wis'niewski J R,Ostasiewicz P,Mann M. J Proteome Res,2011,10:3040
[21] Wis'niewski J R,Zielinska D F,Mann M. Anal Biochem,2011,410:307
[22] Fic E,Kedracka-Krok S,Jankowska U,et al. Electrophoresis,2010,31:3573
[23] Muthurajan R,Shobbar Z S,Jagadish S V K,et al. Mol Biotechnol,2011,48:173
[24] Zhao C F,Wang J Q,Cao M L,et al. Proteomics,2005,5:961
[25] Wei K H,Ying T Y,Hu L P,et al. Short Protocols in Proteomics. Beijing:Chemical Industry Press (魏开华,应天翼,胡良平,等. 蛋白质组学实验技术精编. 北京:化学工业出版社),2010
[26] Islam N,Lonsdale M,Upadhyaya N M,et al. Proteomics,2004,4:1903
