winQTLCart2.0使用程序和QTL分析新方法—关联分析
2014-12-24王竹林刘曙东奚亚军西北农林科技大学农学院陕西杨凌712100
王竹林,刘曙东,奚亚军 (西北农林科技大学农学院,陕西杨凌712100)
作物的许多农艺性状如产量、品质、抗逆性等都是数量性状,由微效多基因控制,这些基因称为数量性状基因(Quantitative Trait Locus,QTL)。传统的数量遗传学把这些微效多基因作为一个整体,用统计学方法分析其总的遗传效应,无法把微效多基因分解为一个个孟德尔因子[1]。分子标记技术的出现和发展为高密度的遗传连锁图谱的构建提供了基础,成为作物各种农艺性状基因分析和定位的重要手段,以分子标记为基础的QTL研究是目前作物遗传育种研究的热点[2]。自PATERSON 等[3]首次应用 RFLP连锁图在番茄中定位QTL之后,国际和国内已有大量的关于QTL研究的报道。QTL研究在分子标记、作图群体和统计分析方法等方面都取得了极大的进展。QTL定位工作现已成为数量性状遗传研究的一种标准程序。QTL研究从一个生育阶段的静态定位发展到全生育期的动态定位,从常规可见的表型发展到生理指标甚至基因的表达水平[1]。利用分子标记进行遗传连锁分析,可将QTL定位,并借助与QTL连锁的分子标记,提高作物育种中对数量性状优良基因型选择的准确性。更为重要的是越来越多的作物包括番茄、水稻、小麦等许多重要性状QTL被分离克隆。笔者介绍QTL分析的原理、软件和最新进展。
1 QTL分析原理、方法和软件
QTL定位的基本原理是分析整个染色体组的标记基因型和数量性状值之间的连锁[4]。QTL定位工作的基本要素包括合适的分离群体,分子标记连锁图以及合适的统计模型与分析方法。QTL定位的一般步骤包括:选择具有目标相对性状的纯系杂交获得作图群体;用大量分子标记检测分离群体,获得分离群体中每一个体的标记基因型;分析群体标记基因型,构建该群体的遗传连锁图;检测分离世代群体中每一个体的数量性状值;在获得了遗传连锁图和分离世代群体中每一个体的数量性状值之后,需要分析标记基因型和数量性状值的关系,确定QTL在染色体上的相对位置。QTL定位的分析方法有方差分析法、区间作图法、复合区间作图、基于最小二乘的复合区间作图和基于混合线形模型的复合区间作图等[2,4]。由于对大量数据进行分析和处理的复杂性,所以必须借助计算机软件才能完成。QTL分析常见软件有QTLnetwork、PLABQTL、MAPMAKER/QTL1.1 、QGene 2.29、QTLMapper2.0 和 winQTLCart2.0 等[1]。
2 QTL分析方法winQTLCart2.0使用程序
2.1 数据准备 准备6个文本数据文件:Chromosome label(连锁群的名称),mark number(每个连锁群上的标记的数目),Mark labels(标记名称),Mark positions(标记之间的距离),Mark genetype(标记基因型),Trait value(性状值),标记基因型从原来的横排转置成竖排,标记顺序按照染色体的顺序。
2.2 运行软件 winQTLCart2.0软件运行时间较长,在运算时不可运行其他大型程序,否则可能引起死机。
打开winQTLCart2.0,点击“new”,出现 Step 1 of 2 对话框,在里面填上相应数据,如该试验连锁群(Cromosome number)为“20”,性状(Trait)为“1”,单株数(Individual number)为“218”,群体(Crossing type)为“SF2”。单株的标记带型记录:“AA”记为“A”,表示该单株包含亲本A的纯合等位基因。“aa”记为“B”,表示该单株包含来自亲本a的纯合等位基因。“Aa”记为“H”,表示该单株是同时携带A和a 2个等位基因的杂合体。“a_”记为“C”表示该单株是A的等位基因的非纯合体(要么是aa基因型,要么是Aa基因型)。“A_”记为“D”表示该单株是a的等位基因的非纯合体(要么是AA基因型,要么是Aa基因型)。“—”表示某单株的数据在该位点缺失(图1)。
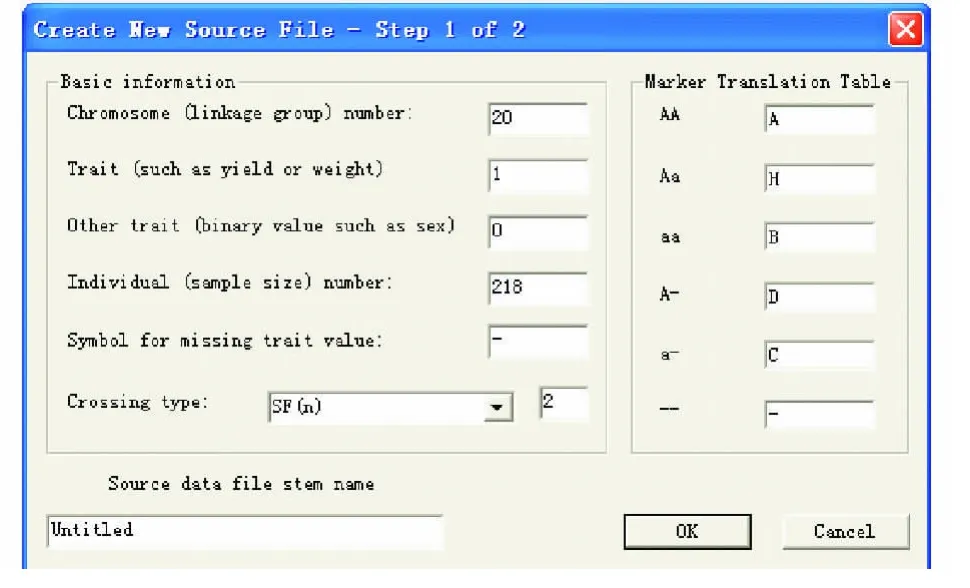
图1 QTL分析群体基本信息
数据输完之后点“ok”,出现“Create New Source File-Step 2 of 2”对话框,点击“Labels”,出现“Data import for new file function”对话框,点击“browse”,将事先准备好的Chromosome label文件输入。按照第一个数据文件的输入程序依次输入剩下的5个数据文件(图2)。
数据输入完之后,点击“ok”按钮,之后产生X.mcd文件,点击QTL分析所要用的方法“IM(区间作图法)”、“CIM(复合区间作图法)”或“MIM(混合复合区间作图法)”。输入分析设定的参数,如LOD值一般设为2.0(图3)。然后点击“start”,分析开始。

图2 QTL分析数据文件输入
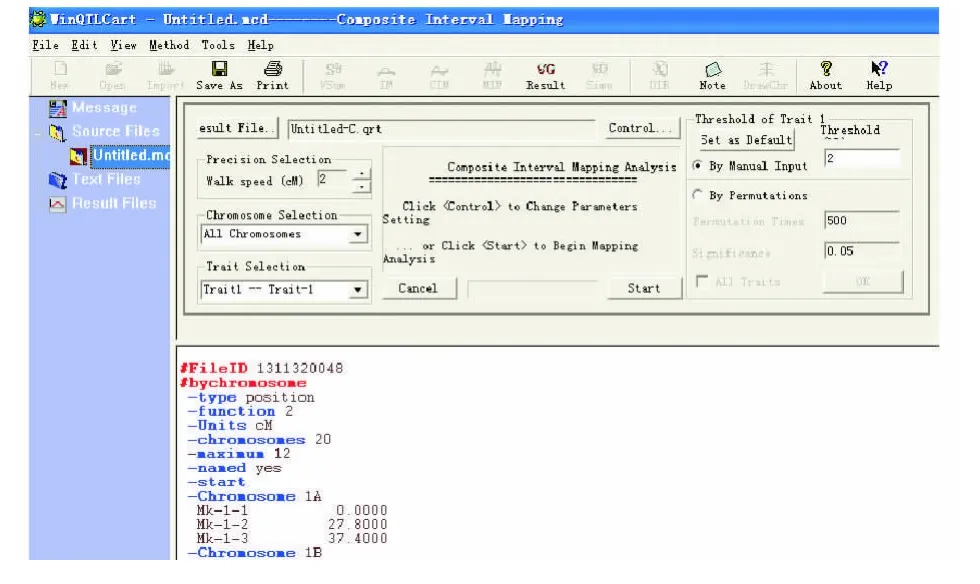
图3 QTL分析基本参数设定
2.3 性状定位 1~2 min运行结束。会出现QTL分析总结文件Untitled-C-eqtl.txt。从此文件能了解QTL定位的结果。如图4所示QTL分析结果为:QTL数目共为4个。第1个QTL位于第4条染色体上,在第2个标记附近,在染色体上的位置是89.81 CM,LOD 为 2.98,加性效应为 -0.950 3,显性效应为0,贡献率为7%等。

图4 QTL分析结果列表
打开软件生成的qrt文件,可以看见曲线图(图5),图上LOD值大于2.0就是QTL位点。按下坐标显示的按钮,将鼠标移动到一个LOD值大于2.0的位置,图标会显示出相关的信息,如第几条染色体、LOD值、距离。
2.4 制图 制图可以用系统自带的画图工具,也可以用photoshop。制图就是注释某个性状QTL在骨架图上的位置,只要根据上一步所确定的QTL在染色体上的位置,用方框、三角、圆圈等不同图形表示不同性状,在骨架图上对应的染色体位置标注上即可(图6)。
3 QTL分析新方法—关联分析
3.1 关联分析在QTL分析中的应用 关联分析(Association analysis)又称连锁不平衡作图(LD mapping)或关联作图(Association mapping),是利用不同基因座等位变异(基 因)间的连锁不平衡关系,进行标记与性状的相关性分析,以达到鉴定特定目标性状基因(或染色体区段)的目的[5]。与传统的连锁分析相比,关联分析具有如下优势:①作图定位更精确。关联分析利用的是自然群体在长期进化中所积累的重组信息,具有较高的解析率,可实现数量性状基因(位点)的精细定位,甚至直接定位到基因本身;②可同时考察一个基因座的多个等位基因。关联分析可实现对其作图群体(自然群体)一个基因座上所有等位基因的考察;③不需构建作图群体。关联分析利用的群体是自然群体,不需再人工构建,省时省力,并有较多的群体可供利用。④可发现较连锁分析更多的QTL位点。鉴于关联分析本身存在的优势,目前已广泛应用于多种作物不同生物特性的研究,如玉米籽粒油分的生物合成[6]、水稻的开花时间和籽粒产量[7]和小麦的籽粒大小和研磨品质[8]等。MACCAFERRI等[9]利用225个分布于21条染色体上的SSR标记对164个硬粒小麦品种的苗期和成株抗叶锈性进行关联分析,鉴定出3个具有较大效应的QTL位点,分别位于7BL末端(Lr14附近)、2A和2B染色体上,其中位于7BL的末端的QTL位点为硬粒小麦中重要的抗病基因,15%的供试品种中都携带该抗病基因位点。张学勇等[10]发现关联分析定位的QTL比以往作图结果丰富,发现了Xgwn149-153-4BL和Xgwm130-132-7AS与粒重显著相关。CROSSA等[11]进一步证明了关联分析能有效地定位和挖掘小麦产量性状基因,在实际育种和种质资源研究中具有重要应用价值。HAO等[12]对266个小麦微核心种质抗赤霉病表型性状和3B染色体3.1-Mb区段42个标记的关联分析结果表明,cfb6059与小麦赤霉病Type II抗性显著相关,且与已广泛应用于抗赤霉病育种的选择标记umn10非常接近。可见关联分析大大提高了目标性状基因或者相关QTL的挖掘和定位,是基因(QTL)定位分析的另一重要方法。
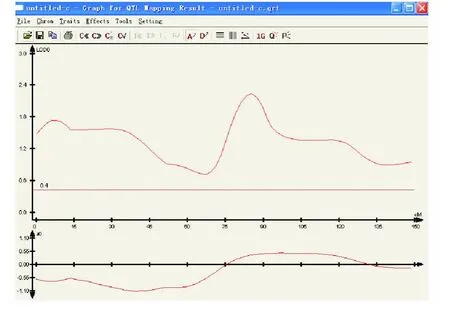
图5 QTL分析结果

图6 QTL在染色体上的位置
3.2 关联分析程序
3.2.1 连锁不平衡分析。
(1)基因型频率统计分析:运用PLINK v1.07软件进行一般的统计分析,包括等位基因频率、标记的杂合率、材料的杂合度等。
(2)连锁不平衡(LD)分析:首先从所有标记中筛选出单个标记缺失率小于10%同时最小等位频率大于10%的标记,然后去除标记间距小于50 Kb的标记,再随机选择几千个标记,进一步进行群体结构分析。用选择的几千个标记,运用Structure V2.3.3软件对500个左右品种或高代品系组成的关联分析群体进行遗传结构分析。根据关联分析群体的遗传组成,将与特定亚群的遗传相似性比例大于60%的材料划分到相应亚群中,而将与任何亚群的遗传相似性比例均小于60%的材料划分到混合亚群中。
(3)邻近LD窗口分析:用TASSEL进行材料间遗传距离计算,并运用Neighbor-joining算法进行聚类图构建。用进化树查看软件FigTree v1.3.1进行画图。
(4)单倍型分析:采用统计算法,以LD为基础,如果2标记存在―强LD,则将该标记划入一种单倍型。单倍型分析用软件PLINK v1.07完成。
3.2.2 关联作图。首先将PLINK生成的单倍型块(Haplotype blocks)结果在Excel中生成可以输入TASSEL的多态性文件格式(如果单倍型中有任何标记缺失,则将该单倍体基因型定为缺失),然后将单倍型数据导入TASSEL对500份左右品种或高代品系的表型数据和SNP数据进行关联作图,找到与目标基因(QTL)紧密连锁的SNP标记,并定位相关基因(QTL)。
4 结语
关联分析较传统QTL分析存在优势,它是建立在自然群体基础之上,可以对一些骨干亲本、大面积推广品种进行全基因组高密度扫描,找到这些重要品种的重要基因组区段,弄清骨干亲本及推广品种的基因组学基础,为分子育种奠定基础。但关联分析也有局限性,如结果受群体结构的影响大,大量数据所导致的统计分析方法的不足以及对遗传多样性低的物种群体作图效果差等。因此,在QTL分析时,应将关联分析与QTL连锁分析相结合,相互补充,相互印证,取长补短,最终实现目标性状的精细作图。
[1]杨绍华.植物QTL 研究进展[J].中国农学通报,2011,27(3):226 -231.
[2]薛永国,刘丽君,高明杰,等.作物中 QTL的研究进展及展望[J].东北农业大学学报,2007,38(4):542 -547.
[3]PATERSON A H,LANDER E S,HEWITT J D,et al Resolution of quantitative traits into Mendelian factors,using a complete linkage map of restriction fragment length polymorphisms[J].Nature,1988,335:721 -726.
[4]章元明.作物QTL定位方法研究进展[J].科学通报,2006(19):2223-2231.
[5]FLINT-GARCIA S A,THORNSBERRY J M,BUCKLER E S.Structure of linkage disequilibrium in plants[J].Annual Review of Plant Biology,2003,54:357 -374.
[6]LI H,PENG Z,YANG X,et al.Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels[J].Nature Genetics,2013,45:43 -50.
[7]HUANG X,ZHAO Y,WEI X,et al.Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm[J].Nature Genetics,2012,44:32 -39.
[8]BRESEGHELLO F,SORRELLS M E.Association mapping of kernel size and milling quality in wheat(Triticum aestivum L.)cultivars[J].Genetics,2006,172:1165 -1177.
[9]MACCAFERRI M,SANGUINETI M C,MANTOVANI P,et al.Association mapping of leaf rust response in durum wheat[J].Molecular Breeding,2010,26:189 -228.
[10]张学勇,童依平,游光霞,等.选择牵连效应分析:发掘重要基因的新思路[J].中国农业科学,2006,39(8):1526 -1535.
[11]CROSSA J,BURGUENO J,DREISIGACKER S,et al.Association analysis of historical wheat germplasm using additive genetic covariance of relatives and population structure[J].Genetics,2007,177:1889 -1913.
[12]HAO C Y,WANG Y Q,HOU J,et al.Association mapping and haplotype analysis of a 3.1-Mb genomic region involved in Fusarium head blight resistance on wheat chromosome 3BS[J].PLOS ONE,2012,7(10):46444.
