无定形态P2O5-V2O5负载型多级孔催化材料的制备及催化合成丙烯酸甲酯
2014-11-18宋伟明田景芝
宋伟明,荆 涛,田景芝
(齐齐哈尔大学化学与化学工程学院,黑龙江 齐齐哈尔 161006)
近年来,以C3 为原料通过羟醛缩合反应合成丙烯酸甲酯成为研究人员广为关注的一种绿色的生产工艺,其中以丙酸为原料合成丙烯酸甲酯,国内外学者进行了一些相关研究[1,2],但是这种方法存在产物收率不高,催化剂容易失活等问题。以C3-醋酸甲酯为原料合成丙烯酸甲酯的过程逐渐引起人们的重视。通过气固相反应,醋酸甲酯与甲醛(或甲缩醛)可以发生羟醛缩合反应生成丙烯酸甲酯,但作为甲醛来源的福尔马林(甲醛水溶液)会引入大量水蒸气,对反应有阻碍作用,同时也会使催化剂随着反应时间的延长而出现失活的现象。Ai[3]以二元氧化物V2O5-P2O5为催化剂,研究了乙酸及其衍生物和甲醛的羟醛缩合反应,发现V2O5-P2O5在反应中具有较高的反应活性,它可以使反应温度下降,而且催化剂中P 和V 原子比和分布是影响催化活性的重要因素。Ai 等[2,4]以丙酸和三聚甲醛为原料,V2O5-P2O5为催化剂,采用固定床反应器对羟醛缩合反应进行了系统研究,发现当催化剂中P 和V 原子比为1.06,丙酸和甲醛物质的量比为2.0 时,选择性可达78%。为了减少活性中心的流失,进一步提高活性中心的分散度并提高催化剂性能,将(VO)2P2O7在少量磷酸作用下负载在二氧化硅表面,制得了P-Si-V 催化剂,其催化活性有了较大的提高。Kitayama 等[5,6]提出了在P-Si-V 催化剂上进行羟醛缩合制丙烯酸(酯)的反应机理:醛与催化剂上的酸中心结合,酸或酸酐与催化剂上的碱中心结合,分别形成中间产物,然后两个中间产物反应生成丙烯酸(酯),即协同作用机理。但P-V 催化剂进行羟醛缩合反应的机理研究尚处于萌芽阶段。谢颖等[7]以V2O5-P2O5/SiO2为催化剂,通过气固相催化羟醛缩合反应,催化甲缩醛与氯乙酸甲酯合成α-氯丙烯酸甲酯,V2O5-P2O5/SiO2催化剂因活性组分不易流失,且转化率和选择性较好,具有一定的实用价值。本工作利用自制的三维有序的大孔/介孔SiO2载体,负载V2O5-P2O5的二元氧化物制备得到易于分离且环境友好的负载型磷钒催化剂,将其用于气固相催化甲缩醛与醋酸甲酯反应生成丙烯酸甲酯。
1 实验部分
1.1 催化剂制备
分别以层析硅胶和三维有序大孔SiO2[8]为载体负载磷钒氧化物,制成负载型磷钒催化剂。将一定量的乳酸加入到适量的二次蒸馏水中制得乳酸溶液,在溶液中加入一定量的NH4VO3并进行磁力搅拌,溶液逐渐变为黄色透明的乳酸氧钒溶液。在体系中加入磷酸溶液,液体变为蓝色透明并逐渐有沉淀生成,继续搅拌2 h。将大孔二氧化硅(或层析硅胶)置于一定量的水中搅拌得到悬浊夜,与磷酸氧钒溶液混合并充分搅拌2 h。待体系进行微波负载一定时间后,放入烘箱中烘干。然后将其置于高温炉中程序升温焙烧,取出研磨至要求的粒径,备用。
1.2 催化剂表征
采用日本岛津/KRATOS 公司AXIS-ULTRA DLD 型X 射线光电子能谱(XPS)仪对催化剂的表面化学组成及价态进行分析,单色铝靶Al(Mono),污染C 峰按284.6 eV 校正,宽谱的通过能为160 eV,步长0.1 eV,功率75 W,窄谱(高分辨)的通过能为20 eV,步长0.1 eV,功率150 W,真空度小于5×10-7Pa。采用日本日立公司的S-4300 扫描电子显微镜(SEM)对样品的形貌进行观测,分辨率0.2 nm(晶格像),加速电压40~120 kV。采用美国康塔仪器有限公司的AUTOSORB-1 全自动物理化学吸附仪进行催化剂的比表面积和孔径分布测定,由BET 方程计算比表面积,孔径分布采用BJH方法计算。采用日本理学工业株式会社的DMAX-IIIB 型X 射线粉末衍射(XRD)仪(Cu 靶,Ka射线,管电压40 kV,管电流30 mA,λ 为0.154 06 nm,扫描范围2θ为5~80°,测定样品的晶体结构。
1.3 催化剂评价
采用固定床微反实验装置对催化剂性能进行评价。以负载型磷钒为催化剂,醋酸甲酯和甲缩醛为原料,在管式反应器中装入催化剂0.55 g,在甲缩醛与醋酸甲酯物质的量之比为1:3,反应温度370 ℃的条件下,反应合成丙烯酸甲酯。用上海科创GC9800 色谱仪对合成产物组成进行定量分析,色谱采用热导池检测器,色谱柱固定相采用天津市化学试剂二厂生产的GDX401,担体为0.180~0.250 mm,用CW3.0 色谱工作站用内标法进行数据处理。
2 结果与讨论
2.1 催化剂磷钒比的选择
在磷钒催化剂催化气相羟醛缩合反应的过程中,催化剂的酸碱平衡在很大程度上决定了催化剂的活性和选择性,而磷钒的相结构决定着催化剂的酸碱活性中心的比例。以层析硅胶为载体,分别制备了活性组分磷钒原子比为1.03 和2.60 的催化剂,记为催化剂1 和催化剂2。
2.1.1 XRD 表征
图1为磷钒催化剂的XRD 谱图。可见:当磷钒原子比(P/V)为1.03 时,在2θ为23.0,26.2 和29.9°处出现(VO)2P2O7晶相所特有的衍射峰,表明在该催化剂表面的活性组分磷钒的晶相主要是(VO)2P2O7;当磷钒原子比为2.60 时,无明显的P 和V 的特征衍射峰,峰强度较小,说明表面活性组分磷钒为无定形态,且分布较均匀。
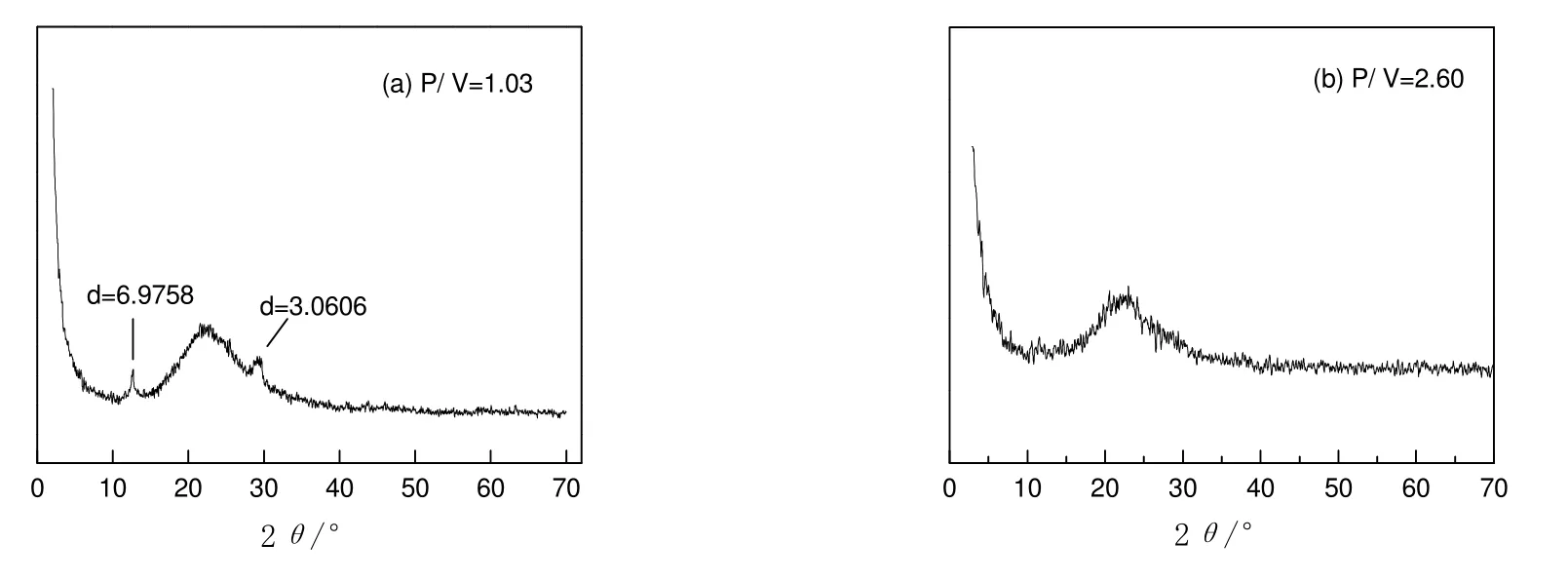
图1 磷钒催化剂的XRD 图谱Fig.1 XRD patterns of V-P catalysts
2.1.2 SEM 表征
图2为以层析硅胶为载体负载不同磷钒原子比催化剂的SEM 照片。由图可知,当负载磷钒原子比为1.03 时,在很多区域都会出现取向性很好的条状晶体,即为(VO)2P2O7晶体,相比于当负载磷钒原子比为2.60 时,负载于层析硅胶载体上具有无定形态的磷钒催化剂,活性组分不能均匀负载于颗粒状的无定形层析硅胶表面,说明以(VO)2P2O7晶体为活性中心的磷钒负载均匀性不是很好,而催化剂2 的活性组分负载均匀。

图2 磷钒催化剂的SEM 照片Fig.2 SEM images of V-P catalysts
2.1.3 催化剂晶体相结构分析
通过能谱分析催化剂1 和2 的表面组成,结果见表1。表1为半定量法能谱分析各组分的含量。由表可知,在催化剂1 中所选择区域的(VO)2P2O7晶体的特征显著,磷钒比为1.05,与所制备的材料的理论值相接近。对催化剂2 测得的V,Si 和P 原子比为1:8:2.57。
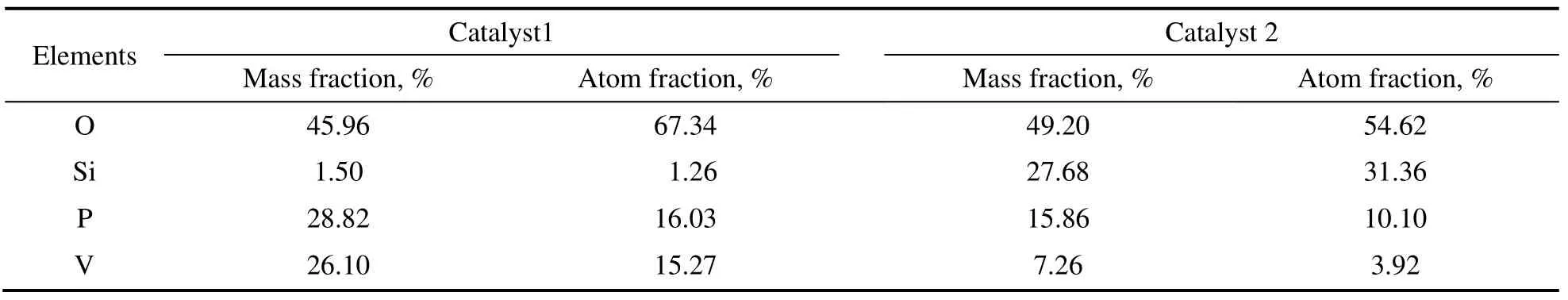
表1 催化剂能谱分析结果Table 1 The analytical results by the energy spectram diagram of catalyst
由文献[9,10]可知,不同磷钒原子比的P2O5-V2O5有着不同的相结构,如表2所示。在催化剂1 中,磷钒原子比为1.03,磷钒的晶相应该是B+β相((VO)2P2O7+VOPO4)。通过XRD 分析可知,催化剂1的相结构以B 相((VO)2P2O7)为主,这说明在体系中V 元素以V4+为主,而且有研究表明,适当的磷过量有稳定B 相的作用[11],而含有V4+的B 相即(VO)2P2O7为主要的催化剂活性物质。在羟醛缩合反应中,碱中心是主要的活性中心,但是具有一定的酸中心,能提高反应的选择性。在以B 相为主的(VO)2P2O7上存在较多的L酸中心和B酸中心,对羟醛缩合反应有促进作用,但只有较纯的V4+物种的(VO)2P2O7催化剂往往催化活性不是很高。如果在催化剂中增加一定量的V5+物种,它会与V4+物种相互作用,使催化剂的活性和选择性大为改善。由此可见,V4+和V5+的平衡对反应很重要,因此,催化剂制备过程中要考虑二者的平衡。
由表2可知,P 和V 原子比在2.0~3.0 属于无定形相。有研究表明[12],无定型相中含有较强的L酸中心和少量的B酸中心,具备促进羟醛缩合反应的要素。Ai[2]的研究表明,V,Si 和P 物质的量之比为1∶8∶2.2的催化剂对羟醛缩合制备甲基丙烯酸甲酯有很好的催化效果。因此,选择磷钒原子比为2.60 的无定形态的磷钒为活性组分进行催化剂的制备。
2.1.4 XPS 表征
通过XPS 宽扫描分别对两种催化剂表面的元素进行分析,结果见图3。可知,样品中含有少量的C,一般认为是测量中带入的C 污染,修正后舍去其峰面积,通过查询标准数据库确定各元素及其组成,结果见表3。由表可知,催化剂1 中磷钒原子比为1.03,催化剂2 中为2.60,与表1的分析结果相近。由表3中各元素的电子结合能可看出,催化剂1 中V 元素的电子结合能与文献报道的(VO)2P2O7的相应元素结合能数值516.9 eV 接近,这表明催化剂1 中V 元素主要以(VO)2P2O7形式存在。比较表3中催化剂1 与2 中的各元素的结合能可看出,两种催化剂的Si2p,P2p和O1s 的电子结合能非常接近,但是V2p3/2 的结合能相差较大,这表明两种催化剂中V 物种的种类明显不同。

表2 P2O5-V2O5 的相结构Table 2 Phase structure of P2O5-V2O5
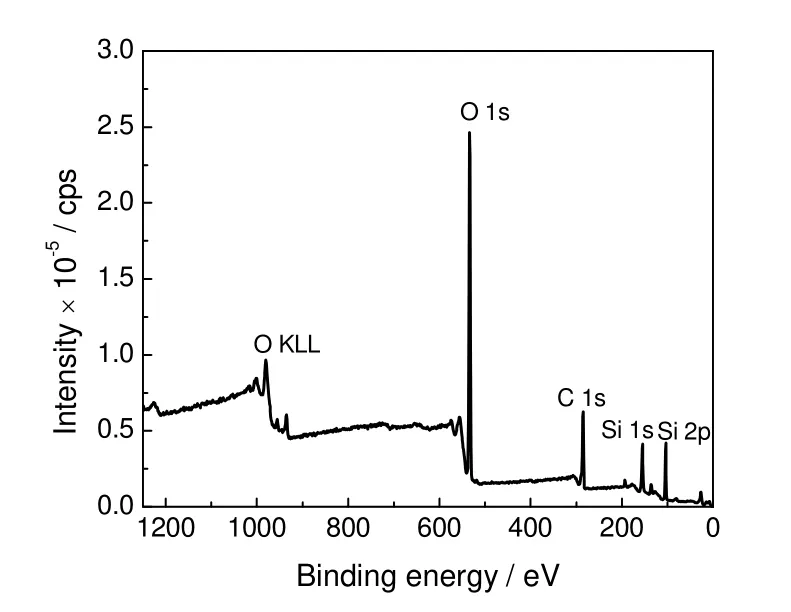
图3 磷钒催化剂的XPS 宽扫描图谱Fig.3 XPS wide scanning spectra of V-P catalyst
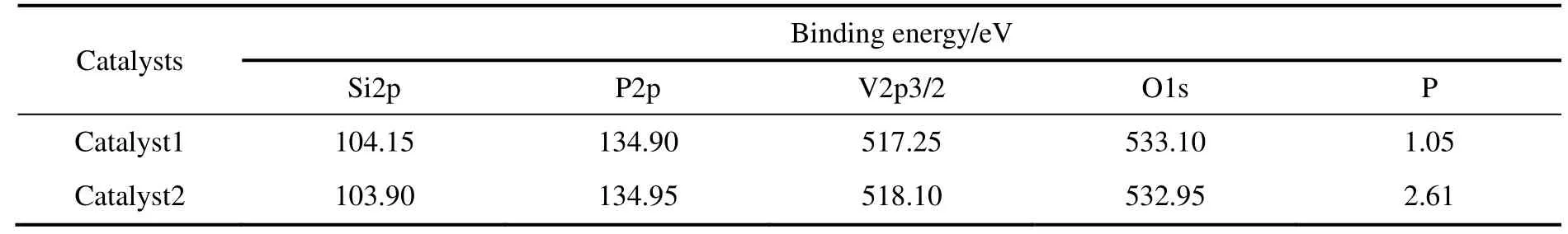
表3 XPS 分析催化剂中各元素结合能数据Table 3 Binding energy of elements on the surface of different catalysts
为了进一步了解两种催化剂表面V 物种的分布情况,对两个样品的V2p3/2 特征峰进行了分峰拟合(取V4+的V2p3/2 结合能为516.6 eV,V5+的V2p3/2 结合能为517.7 eV[13]),估算催化剂表面V4+和V5+两种物种的比例,结果见图4和表4。

图4 磷钒催化剂的V2p3/2 分峰拟合Fig.4 V2p3/2 curve fittings of V-P catalyst
由拟合结果可看出,催化剂1 中磷钒是以V4+物种占绝大多数,其原子百分含量为71.26%,是以(VO)2P2O7为主的催化剂。催化剂2 中的活性组分是无定型相的磷钒,V 以V4+和V5+两物种存在,而且V5+物种含量明显增加到47.24%。在设计的活性组分磷钒比为2.6 的催化剂中,磷钒相结构为无定型相,在V4+基础上明显增加了V5+的比例,有助于促进V4+和V5+的协同作用,有利于羟醛缩合反应的进行。

表4 V2p3/2 分峰拟合结果Table 4 The curve fittings results of V2p3/2

图5 大孔二氧化硅为载体的磷钒催化剂SEM 照片Fig.5 SEM image of V-P catalysts supported on macroporous silica
2.2 催化剂载体的选择
2.2.1 大孔SiO2为载体的磷钒催化剂的微观形貌
以大孔SiO2为载体,磷钒原子比为2.60 制备的磷钒催化剂3 的SEM 照片见图5。从图5可看出,催化剂整体形貌的有序性,且大孔孔道通透,大孔之间的孔壁规整,但由于大孔负载活性组分使得其局部会产生少量缺陷,孔间存在大量的介孔孔窗。从图5还可以看出,催化剂具有三维有序笼状结构,呈大孔/介孔形态。具有无定形态的磷钒活性组分主要负载于载体孔壁上,且负载均匀。
2.2.2 负载型磷钒催化剂的比表面积及孔径分析
采用N2吸脱附对催化剂2(层析硅胶为载体,磷钒原子比为2.60)进行微观结构表征。等温曲线及孔径分布见图6。图6(a)为Ⅳ型吸附等温线。在相对压力(P/P0)小于0.2 处有一微小平滑拐点,说明存在微孔;P/P0在0.6~0.9 时出现明显滞后环,说明在N2吸附与脱附过程中发生毛细管凝聚现象。由图6(b)孔径分布曲线可知,磷钒催化剂的孔径分布主要集中在2.5~7.0 nm。其比表面积为238.515 m2/g,在P/P0为0.99 时,催化剂2 的孔容为0.496 cm3/g。

图6 催化剂2 的吸附脱附等温线和孔径分布Fig.6 The adsorption-desorption isotherm and pore distribution of catalyst 2
采用N2吸附法和压汞法对催化剂3 进行比表面积及孔结构分析。图7为SiO2的N2吸附脱附等温线。由图可知:当P/P0较小时,没有明显的拐点存在,说明所制备的SiO2无微孔或微孔较少,可以快速地达到单分子层饱和吸附。随着相对压力P/P0增大,吸附量缓慢增加,说明发生了多分子层吸附;当P/P0大于0.85 时,吸附量急剧上升,并出现滞后环,表明孔中有凝聚现象发生,且存在介孔。由BET 方法计算得出,材料的比表面积为110.2 m2/g。采用BJH 法计算得到,当P/P0为0.99 时,孔容为3.05 cm3/g。
大孔SiO2的BJH 脱附曲线孔径分布如图8所示。从图可知,孔径分布集中在中孔5.0 nm 处,由此可推测出其为小孔窗及中孔孔壁的空腔所形成的,但是,在BJH 吸附孔径分布曲线图中,5.0 nm处无此尖峰存在,说明此处峰可能为假峰,而在40~60 nm 有一较宽的峰带,说明介孔孔窗的存在。
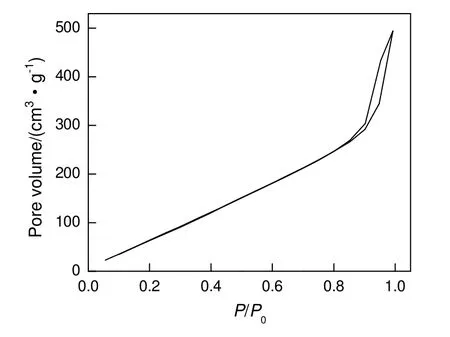
图7 大孔SiO2 的N2 吸附-脱附等温线Fig.7 N2 adsorption-desorption curve of macroporous SiO2

图8 大孔SiO2 的孔径分布Fig.8 Pore size distribution of macroporous SiO2
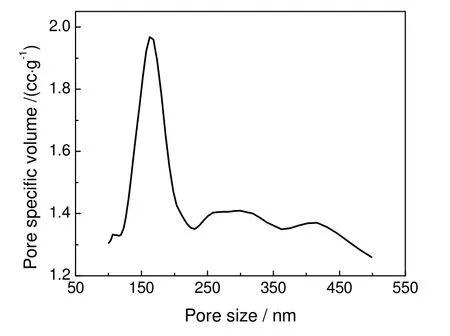
图9 压汞法大孔SiO2 孔径分布曲线Fig.9 Distribution curve of macroporous SiO2 by mercury porosimetry method
图9为压汞法测得的大孔SiO2孔径分布曲线。由图可知:在160 nm 左右有一个尖峰,证明其最可几孔径为163 nm,孔分布较窄;孔道集中在100~200 nm,在200~500 nm 存在两个脱尾峰,这说明样品中存在200~500 nm 的孔径分布。这可能是由于采用高压压汞测定时,样品在较高压力下孔结构受损所至。测定结果采用韦斯布恩方程统计得出孔容为3.19 cm3/g
优良的载体固然要求具有较大的比表面积,但通透的孔道以及充足的反应空间对催化反应来说也是必要的,这样才可发挥出良好的催化性能。制备的以大孔二氧化硅为载体的磷钒催化剂比表面积要小于层析硅胶为载体的催化剂,但具有均匀有序而通透的大孔/介孔多级孔道,大孔孔腔提供了优良的反应空间,而且使磷钒比为2.60 的无定型态的活性组分更适合于均匀负载在大孔二氧化硅的表面,无论从孔结构还是磷钒的相结构上都建立了良好的微观环境,从而构成了具有对羟醛缩合有良好催化性能的要件。
2.3 不同载体对催化性能的影响
不同催化剂载体对反应催化性能的影响见表5。由表可知,以大孔SiO2为载体的催化剂,其甲缩醛的转化率、丙烯酸甲酯的选择性和收率比以层析硅胶为载体的催化剂都要高。这是由于大孔SiO2为载体的磷钒催化剂,其孔道通透,催化剂活性组分在其表面负载均匀,同时具有较大的比表面积,既使得原料可以在催化剂表面反应比较充分,同时又有效地减少了原料在催化剂表面的停留时间,减少了副反应的发生。虽然层析硅胶具有较大的比表面积,但其颗粒较小,没有通透的孔道,催化剂装填后易使催化剂床层中阻力增大,使得原料在催化剂床层中停留时间过长。

表5 不同载体催化剂对催化性能的影响Table 5 Effects of different catalyst supports on catalytic performance
3 结 论
以三维有序大孔二氧化硅为载体,负载具有无定形态的磷钒活性组分的催化剂具有三维有序笼状大孔结构,大孔间有大量介孔孔窗存在,且活性中心在催化剂上负载均匀。磷钒比为2.6 的催化剂,其活性组分呈无定形相,其中V 同时以V4+和V5+两物种存在。在磷钒原子比为2.6 的情况下,以大孔SiO2为载体的P2O5-V2O5催化剂比层析硅胶为载体的催化剂具有更好的催化性能。
[1]Tai J R, Davis R J.Synthesis of methacrylic acid by aldol condensation of propionic acid with formaldehyde over acid-base bifunctional catalysts[J].Catalysis Today, 2007, 12(4):42-49.
[2]Ai M, Fujihashi H, Hosoi S, et al.Production of methacrylic acid by vapor-phase aldol condensation of propionic acid with formaldehyde over silica-supported metal phosphate catalysts[J].Applied Catalysis A:General, 2003, 252(1):185-191.
[3]Ai M.Vapor-phase aldol condensation of formaldehyde with acetic acid on V2O5-P2O5catalysts[J].Journal of Catalysis, 1987, 107(1):201-208.
[4]Ai M.Reaction of propionic acid with methylal over vanadium-silicon-phos phorus oxide[J].Applied Catalysis, 1990, 63(1):365-373.
[5]Kitayama Y, Michishita A.Catalytic activity of fibrous clay mineral sepiolite for butadiene formation from ethanol[J].Chemical Communications, 1981, (9):401-402.
[6]Suzuki E, Idemura S, Ono Y.Catalytic conversion of 2-propanol and ethanol over synthetic hectorite and its analogues[J].Applied Clay Science, 1988, 3(2):123-134.
[7]谢 颖, 肖林久, 王宜阳, 等.气固相催化羟醛缩合合成α-氯丙烯酸甲酯的研究[J].辽宁化工, 2005, 34(5):203-205.Xie Ying, Xiao Linjiu, Wang Yiyang, et al.Study on synthesis of alpha-methyl chloroacrylate by the vapor-phase aldol condensation[J].Liaoning Chemical Industry, 2005, 34(5):203-205.
[8]邬泉周, 何建峰, 李玉光.聚苯乙烯胶晶及三维有序大孔SiO2膜的制备及表征[J].化学研究, 2008, 19(3):5-8.Wu Quanzhou, He Jianfeng, Li Yuguang.Preparation and characterization of polystyrene colloidal crystal films and ordered macroporous silica films[J].Chemical Research, 2008, 19(3):5-8.
[9]Bordes E, Courtine P.Some selectivity criteria in mild oxidation catalysis:VPO phases in butene oxidation to maleic anhydride[J].Journal of Catalysis, 1979, 57(2):236-252.
[10]Moser T P, Schrader G L.Selective oxidation ofn-butane to maleic anhydride by model VPO catalysts[J].Journal of Catalysis, 1985,92(2):216-231.
[11]韩维屏, 邓启刚.金属原子对磷钒催化剂中未知相与酸性的影响[J].石油学报(石油加工), 1990, 6(1):53-59.Han Weiping, Deng Qigang.Effect of the promoter metal atoms on the unknown phase and the acidity of P2O5-V2O5catalyst[J].Acta Petrolei Sinica(Petroleum Processing Section), 1990, 6(1):53-59.
[12]周 艳, 韩维屏, 相 卫.P2O5-V2O5的XRD、IR 及ESR 研究[J].石油化工, 1987, 16(2):76-80.Zhou Yan, Han Weiping, Xiang Wei.Investigation of P2O5-V2O5catalyst for oxidation ofn-butane to maleic anhydride[J].Petrochemical Technology, 1987, 16(2):76-80.
[13]Abon M, Bere K E, Tuel A.Evolution of a VPO catalyst inn-butane oxidation reaction during the activation time[J].Journal of Catalysis,1995, 156(1):28-36.
