大气颗粒物中有机物色谱分析的样品制备技术
2014-10-22吴大朋关亚风
郝 亮,吴大朋,关亚风*
(1.中国科学院大连化学物理研究所,辽宁 大连 116023;2.中国科学院大学,北京 100039)
大气颗粒物是指悬浮在大气中的固体或液体颗粒[1]。按粒径(空气动力学直径)可将大气颗粒物分为总悬浮颗粒物(total suspended particles,TSP;空气动力学直径Dp≤100μm)和可吸入颗粒物(inhalable particles,IP;Dp≤10μm,也称PM10)。通常所说的大气细颗粒是指大气中空气动力学直径小于2.5 μm(Dp<2.5μm)的颗粒物,也称PM2.5。由于粒径小,在空气中漂浮时间长,PM2.5可携带表面吸附的有毒有害物质由呼吸系统进入人体肺部,在肺部沉积或通过肺泡进入人体血液循环,对人类健康危害极大[2],因而引起社会的广泛关注。
颗粒物的来源有两类[3]:一类是各种源的直接排放,即一次源;另一类是大气中的化学过程使原来的气态组分氧化而产生的粒子,即二次源。人类活动(如工厂排放、汽车尾气排放等)和自然来源(如火山喷发、植被排放等)对颗粒物的两种来源都有贡献。由于来源及形成过程不同,使得颗粒物的平均组成随粒径、时间、地点以及单一颗粒物的总组成的变化而发生明显的变化。颗粒物的化学组成包括各种微量金属、无机氧化物、硫酸盐、硝酸盐和有机化合物等。有机物是大气颗粒物重要的物质基础,在颗粒物组成中占有很大比例(通常占大气细颗粒总干重的10%~70%),但由于其种类繁多,分析复杂,仅有10%~20%的有机物得到了定性和定量分析。表1[3,4]列出了已被测出或由光化学和热力学计算预测到的有机化合物种类,主要包括烷烃、烯烃、多环芳烃等。其中多种有机物如多环芳烃、多氯联苯等具有很强的生理毒性以及致癌致畸性,是颗粒物生理环境毒性的主要来源。此外,有机物的存在也对颗粒物的吸湿性有重要的影响,而吸湿性影响颗粒物大小进而影响大气能见度,也影响着颗粒物作为云凝结核的性质,从而影响气候变化。因此,大气颗粒物中有机物成分的分析对深入研究大气颗粒物对人类健康、能见度以及气候的影响,解析气溶胶来源,制定PM2.5控制相关法规以及风险管理方法具有重要意义[5]。

表1 大气颗粒物中的有机组分[3]Table 1 Organic constituents of atmospheric particulate matter[3]
由于颗粒物中有机物种类繁多且基质复杂,分离和鉴定是分析的关键。目前,色谱以及色谱-质谱联用是大气细颗粒有机物分离鉴定的主要手段。在分析前需要将样品转化为适合色谱分析的形式,也即需要进行样品制备。对于成熟的色谱分析方法来说,样品制备已成为分析速度以及分析误差的决定步骤,在分析方法中尤为重要。本文对近年来大气颗粒物中复杂有机物成分色谱分析前的样品制备方法研究进展作一总结,并简要介绍了各种方法的优缺点。
1 大气颗粒物有机物色谱分析的样品制备方法
颗粒物中有机物分析的一般步骤为:用玻璃(或石英)纤维滤膜或铝箔收集颗粒物样品,采用适当的样品前处理方法提取样品,将提取物引入色谱系统分离后,用适当的检测器检测。根据样品制备原理的不同,可分为溶剂提取(solvent extraction,SE)和热解吸(thermal desorption,TD)两大类。
1.1 溶剂提取
溶剂提取是采用溶剂溶解的原理进行有机物的提取。图1为颗粒物溶剂提取的过程总结,主要方法有索氏提取[6]、超声辅助提取[7,8]、微波辅助提取[9,10]、溶剂加压提取[11-14]、超临界流体提取[15,16]、固相微萃取、中空纤维液相微萃取等。表2是对几种溶剂提取方法在大气颗粒物样品分析中的应用实例及方法的优缺点总结。提取溶剂一般根据“相似相溶原理”进行选择。提取后根据分析对象的不同还需要对提取液进行净化、浓缩等处理。
1.1.1 索氏提取
索氏提取(Soxhlet extraction)是最经典的有机物提取方法。虽然存在着提取时间长,有机溶剂用量大等缺点,但索氏提取能提取完全,选择性和可行性都较好,因此仍被用来开发新的分析方法及作为标准方法与一些新的提取方法进行比较。
1.1.2 超声辅助提取
索氏提取耗时长,且同时处理的样品数量少。为缩短提取时间,增加样品处理通量,超声常用于辅助提取。超声辅助提取所用的有机溶剂量较索氏提取有所减少,且提取时间大幅缩短,并可同时处理多个样品,因此在气溶胶样品分析中得到广泛的应用。
1.1.3 微波辅助提取
微波辅助提取是一种利用微波加速样品提取的技术。样品放入微波提取池中,加入溶剂后用微波辐射,具有处理时间短,选择性较好的优点。
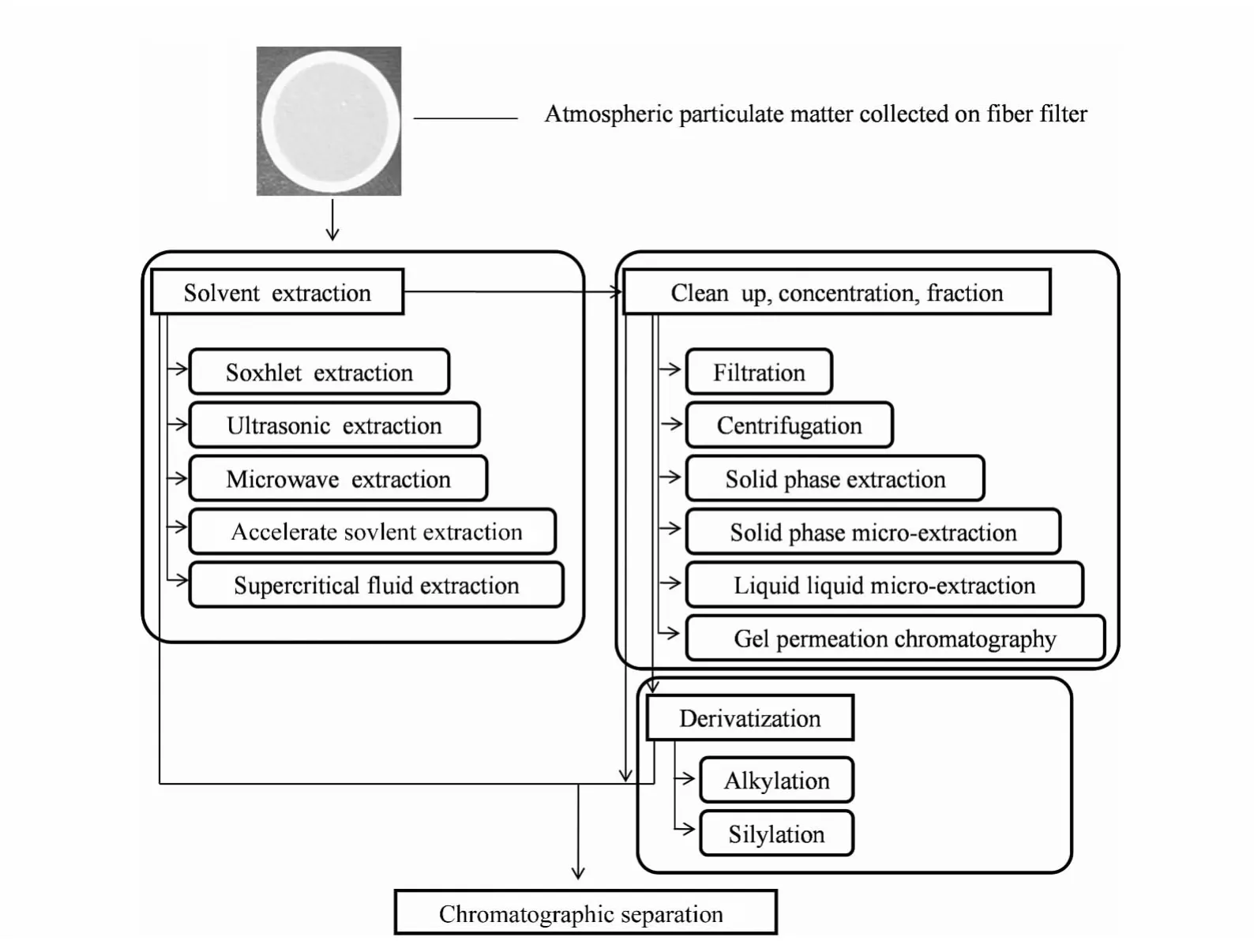
图1 大气颗粒物溶剂提取流程Fig.1 Solvent extraction process for analysis of atmospheric particulate matter

表2 大气颗粒物溶剂提取方法比较Table 2 Comparison of different solvent extraction methods for analysis of PM samples
1.1.4 加速溶剂提取
加速溶剂提取是在较高温度和压力下用液态溶剂对样品进行提取的方法。由于有机溶剂用量少、提取效率高、提取时间短,加速溶剂提取应用较广。Schantz等[11]用4种 NIST(美国 National Institute of Standards and Technology)的标准参考材料(standard reference material,SRM)考察了溶剂、静态循环次数及时间、压力以及温度对多环芳烃及硝化多环芳烃提取的影响。结果表明:二氯甲烷/甲苯与甲苯/甲醇混合液的提取效率基本相当;3~5次循环、每次循环5~30 min对提取效率没有明显的影响;萃取压力为13.8~20.7 MPa,提取效率没有明显变化;200℃时PAHs提取效率较100℃时高。
常规加压溶剂萃取池的池体积最小为11 mL,提取时需要消耗的样品和提取溶剂仍然较多,样品利用率低。为解决这一问题,关亚风课题组[13]对加速溶剂萃取装置的小型化进行了探索,开发了微池加压溶剂萃取仪,并利用该装置建立了小型加压溶剂萃取-离线气相色谱分析方法,证明了加压溶剂萃取小型化的可行性。与超声萃取相比,加压溶剂萃取的萃取效率和萃取时间都有明显的优势。结合毛细管气相色谱大体积进样技术,该课题组[14]又报道了微池加压溶剂提取-大体积进样-气相色谱在线联用测定PM10中多环芳烃的分析方法。该方法将全部萃取液一次转入毛细管气相色谱,使检测灵敏度得到极大提高。使用小体积采样器,只需采集1 h的大气细颗粒样品就能定量分析大气颗粒物中的有机物,从而可以反映大气细颗粒短时间的化学变化。
1.1.5 超临界流体提取
由于有机溶剂造成二次污染,以低毒的CO2为溶剂的超临界流体提取(SFE)也被用于颗粒物中有机物的提取。超临界流体提取具有成本低、耗时短、易与其他分析技术联用的特点[15],可提取很多有机物甚至包括相对分子质量高和中等极性的化合物,是一种具有选择性的提取技术。由于CO2极性很低,不适合于强极性有机物的提取。为了提高溶剂的溶解能力和选择性,可以加入非极性或极性溶剂。
1.1.6 净化、浓缩及衍生方法
由于大气颗粒物中有机物组成十分复杂,单种有机物含量极低,各种提取方法通常需要多种溶剂多次提取,合并后的提取液体积较多,因此不适合直接进行色谱分析[17]。因此,在样品提取后需要对提取液进行适当的浓缩、净化、预分离等处理以减少基质干扰,提高目标分析物在提取液中的浓度。合并后的提取液首先用旋转蒸发仪将体积减少到几毫升,如需更换溶剂或进一步浓缩,可以使用超纯氮或氦气减小体积至1μL以下;也可使用K-D浓缩仪蒸发提取物。提取液中可能含有来自气溶胶样品的固体颗粒或来自滤膜的固体碎片,可采用过滤、离心或固相萃取等方法除去。净化和预分离通常采用固相萃取(SPE)完成,将提取液通过一根固相萃取柱,然后用不同极性的溶剂淋洗,收集流出的组分。当目标分析物为极性化合物或沸点较高时,采用气相色谱分析时还需对样品进行衍生化。如有机酸用重氮甲烷或三氟化硼醇溶液衍生为酯,醇用硅烷化试剂衍生。净化和浓缩使得样品前处理繁琐费时,且容易引入误差,造成分析结果不准确。
近年来,固相微萃取以及液液微萃取技术的应用使得净化和浓缩可同时进行。van Pinxteren等[18]报道了三相中空纤维液相微萃取富集颗粒物中一元羧酸、二元羧酸等水溶性有机物方法。方法以含10% (w/v)三辛基磷酸的二己基酯为支撑液膜,颗粒物水提液作为提取相,氨水溶液作为受体相,振荡提取2 h,大多数化合物得到较为满意的回收率和重复性。Menezes等[19]报道了用纯水、二氯甲烷和丙酮混合液在超声浴中对采集有气溶胶样品的滤膜平衡提取5 min,再用100μm直径的经过制冷的聚二甲基硅氧烷(PDMS)萃取纤维对提取液中的PAHs进行净化浓缩,GC/MS分析大气气溶胶中PAHs的分析方法。对NIST SRM1649b标准参考材料的分析结果表明,方法对样品中的PAHs具有高的准确度和精密度,以及低的检出限和定量限。此外,采用吸附搅拌棒提取的技术也有报道[20]。
1.2 热解吸
溶剂提取的主要优点在于有不同极性的提取溶剂可供选择,适应提取不同极性的目标分析物,且一般都具有较高的提取效率。因此,虽然溶剂提取存在着提取时间长、提取过程繁琐、容易引入分析误差、样品利用率低等缺点,但在提取大气颗粒物中极性有机物(如左旋葡聚糖、多环芳烃衍生物、有机酸)时仍是不可取代的提取方法。对大气颗粒物中非极性有机物的提取,热解吸是可供选择的另一类方法,近年来已被广泛用于大气颗粒物中非极性有机化合物的提取。对其全面详细的介绍可参考Hays等[5]和Chow等[21]的综述。热解吸是指通过加热将样品中的挥发性有机物提取出来,图2为大气颗粒物热解吸所用装置的结构示意图。根据提取过程中所使用的加热设备,热解吸可分为单炉加热、冷阱聚焦的双加热炉体系、进样口直接引入热解吸3种形式。
1.2.1 单炉加热
单炉加热热解吸方式[22-24]是将采样管或装有样品的热解吸管放在GC进样口上方的加热炉中加热解吸。加热炉通常为一铝块,铝块中嵌入加热棒用于加热,热电偶用于控温。载气携带加热解吸出的有机物气体通过传输线直接送入冷却的毛细管柱头聚焦,待加热解吸完成后启动色谱仪开始分析。在国内,中科院大连化学物理研究所关亚风课题组[25]构建了一种直热式热解吸装置(TDU)。该装置升温速率快,采用载气吹扫设计避免了柱外死体积和样品残留问题。TDU能直接安装在气相色谱进样口上,与气相色谱或气相色谱-质谱联用实现各级大气颗粒物中痕量半挥发性有机物的定性与定量分析。使用这一装置对收集了PM10的样品滤膜或铝箔直接加热,同时以3 mL/min的解吸气流将解吸的化合物送入40℃的色谱柱柱头,实现对解吸化合物的聚焦。在310℃热解吸温度下,持续解吸进样10 min,气相色谱分析没有明显的谱带展宽,得到了很好的分离结果。

图2 颗粒物热解吸装置的结构示意图Fig.2 Conceptual diagrams of different thermal desorption-gas chromatographic approaches
1.2.2 冷阱聚焦的双加热炉体系
在单炉加热的热解吸装置后串联一个冷阱构成了有冷阱聚焦的双加热炉体系,冷阱中的衬管填装石英棉或玻璃珠等惰性材料,从第一级加热炉解吸出的有机物完全通过冷阱聚焦在填装的惰性材料上。与单炉加热相比,冷阱聚焦装置可允许使用较高流速的热解吸载气。在单炉加热装置中,由于热解吸气直接通过传输线送入柱头聚焦,太高的热解吸气流速会破坏质谱的真空度,通常使用的热解吸气流速为3 mL/min左右。有冷阱的二级解吸装置在热解吸过程中使用的解吸气流速通常为150~300 mL/min。高的热解吸气流速更有利于样品中的半挥发性有机物从样品基质中快速挥发出来,降低热不稳定化合物分解的可能性。此外,热解吸时间很长,通过冷阱聚焦后进样,低沸点化合物也能得到很窄的进样谱带。Waterman等[26]最早报道了带有冷阱聚焦的热解吸体系对NIST标准参考材料(SRM1649a)进行了分析。目前,具有冷阱聚焦的热解吸系统已经商品化,文献[27-30]报道的大多数气溶胶热解吸分析都采用这种方式。Ding等[28]使用商品化的热解吸器和安捷伦GC6890/MS5973系统对大气颗粒物中有机物进行了分析;Gil-Molto等[27]用自动热解吸系统,用传输线(152 mm×0.70 mm o.d.×0.53 mm i.d.)连接商品化热解吸器(Gerstel TDS2/TDSA,Germany)和冷阱冷却程序升温进样系统(Gerstel CIS4),对PM10和PM2.5中的多环芳烃(PAHs)进行了分析,结果表明使用这种体系可直接对大气颗粒物中的有机组分进行热解吸色谱分析。
1.2.3 进样口直接引入热解吸
具有外部加热炉的热提取方法需要一根传输线和接头连接解吸器和GC进样口。因此,传输线、冷点可能会造成有机物吸附损失。最简单的热解吸方法是在GC进样口进行热解吸。因此,Falkovich等[31]对气相色谱进样口做了微小改动,使气溶胶样品滤膜可以放入一个小样品瓶中,再将样品瓶放入进样口中对样品进行分析。Ho和Yu等[32-34]将气溶胶样品滤膜放入热解吸衬管中,再直接放入冷却的气相色谱程序升温进样口中,隔垫密封后进样口升温,解吸后的分析物聚焦在30℃的色谱柱头,解吸完成即开始色谱分析。
1.2.4 热解吸过程中的一些问题
TD结合GC-MS技术已被广泛应用于大气颗粒物中痕量半挥发有机物的测定。同溶剂萃取方法相比,TD具有许多优越性[5]:TD技术不使用有机溶剂,无溶剂中杂质的污染;操作简单,可在线处理样品,样品利用率接近100%,减少了样品用量从而缩短了颗粒物采样时间。但TD技术也存在着一些问题。在解吸过程中,一些细颗粒可能被解吸气流从采样滤膜上吹下,如果它们被载气带入GC-MS系统,颗粒物可能在进样口沉积进入色谱柱,甚至进入MS系统。随着颗粒物的积累,精密的GC-MS系统很快会出现故障,在进行几轮TD-GC分析循环后,色谱柱也会发生退化。除硬件损伤外,也有可能发生分析物损失、背景信号升高、峰展宽、峰重叠等现象。因此在解吸气引入分析系统之前,必须尽可能除去悬浮的颗粒物。为解决这一问题,通常的做法是向热解吸衬管中填装去活玻璃纤维棉。然而,对于颗粒物的移除效率以及去活玻璃纤维棉填装参数等过滤条件却未有报道。最近,中科院大连化学物理研究所关亚风课题组对这一问题进行了系统的研究,采用颗粒物过滤理论对TD过程中最易穿透的颗粒物粒径、拦截颗粒物所需填装的玻璃纤维棉等填装参数进行了计算;进一步又设计实验对计算结果进行了验证。结果表明:在通常的TD条件(解吸温度300℃,解吸气流速0.4~4 cm/s)下,向热解吸衬管中填装直径为5μm的去活玻璃纤维棉,填充率0.039,填充厚度30 mm可以完全除去热解吸气流中的颗粒物。
此外,在TD过程中还会出现残留、传输损失、分析物转换以及降解等可能干扰有机物分析的问题。这些问题都与样品中极性官能团(如羧酸和醇)有关。极性官能团吸附在分析仪器的表面(如GC进样口)会导致分析物解吸不完全;在较高温度下极性化合物容易发生转化和降解[21]。在TD分析中,羧酸和烷醇的回收率一般低于30%。这些化合物出现在气溶胶中可能会导致其他化合物定性错误。例如Williams等[35]虽定性分析了极性的左旋葡聚糖,但它的定量限高,标准曲线回归值低,无法得到满意的定量结果。高极性化合物如羧酸,虽然它们的沸点比解吸温度低,在可接受的分析不确定度下,尚没有热解吸提取定量分析的方法。针对这类化合物,有研究者提出了衍生热解吸的处理方法。Beiner等[36]以四甲基氢氧化铵为衍生试剂,对颗粒物进行衍生后进行热解吸,建立了颗粒物中有机酸的定量分析方法;另外,Sheesley等[37]用叠氮化钠处理滤膜样品,建立了羧酸原位甲基化热解吸的方法;Orasche等[38]报道了一种原位衍生热解吸,一次性测定颗粒物中极性和非极性有机化合物的分析方法,该方法能对大气颗粒物中的羟基和羧基化合物如脱水糖、醇、酚、酸等有机物衍生后分析。衍生在热解吸过程中完成,分析时间没有延长。目前,这类方法仍处于研究阶段。
1.3 溶剂提取与热解吸的比较
SE-GC/MS以及TD-GC/MS分析大气颗粒物有机组分的对比研究报道很多。Greaves等[22]用一个大体积采样装置和小体积热解吸采样管平行采样,大体积采样器采集的样品用二氯甲烷超声搅拌进行溶剂提取。比较定量结果表明,对大多数多环芳烃和正构烷烃,TD的分析结果与SE的分析结果的比值在2.5倍以内。一半的化合物TD的分析结果与SE的分析结果比值在1.25倍以内。其中小体积采样热解吸分析大气颗粒物中多环芳烃和正构烷烃的浓度范围分别是0.06~7.8 ng/m3和0.3~15 ng/m3。溶剂提取后的滤膜接着热解吸仍能提取相当于溶剂提取量5%~10%的多环芳烃和约20%的正构烷烃,这一结果表明热解吸能更有效地提取多环芳烃和正构烷烃类化合物。Jeon等[39]以及 Ho和Yu[34]也分别比较了SE-GC/MS和TDGC/MS分析PAHs和正构烷烃的结果。两种方法对大多数PAHs和正构烷烃分析结果的比值在2.5倍以内。Jeon等证明SE-GC/MS和TD-GC/MS的分析结果相关性很好,除正构烷酸(R2=0.731)外,藿烷和甾烷(R2=0.998)、多环芳烃(R2=0.978)、正构烷烃(R2=0.975)以及酚类(R2=0.940)都有较好的相关系数。Hansen等[40]比较了超临界流体提取和热提取分析大气气溶胶样品和空白基质加标样品。同超临界流体提取相比,醇(C13~C28)以及羧酸(C9~C20)的热提取回收率很差(<30%)。质谱分析表明:醇分子发生了脱水的热化学裂解,生成烯烃;C9~C20的羧酸在热提取过程中的脱水也很显著。Neususs等[41]证明:除了用热解吸分析的茚并[1,2,3]芘和苯并[g,h,i]芘的浓度更低以外,SE-GC/MS和 TD-GC/MS分 析 PAHs结 果 在130%±30%范围内一致。
2 结论
大气颗粒物中的有机组分分析对人类了解颗粒物的危害,研究颗粒的形成,追踪其来源具有重要意义。由于颗粒物组成复杂,现有的分析监测水平只能测定样品中所有有机物的一小部分。样品前处理对色谱分析方法至关重要。溶剂提取方法适合于不同极性化合物的提取,但样品利用率低,颗粒物样品需要量大,且提取液预分离、浓缩与分析测定基本上是离线进行的,使得样品的分析时间长,成分损失和变化较大。另外,有机溶剂用量大,对分析人员和环境的危害也大。微池加压溶剂提取为样品前处理与色谱分析的在线联用,以及开发高灵敏选择性的分析方法提供了方向。热解吸方法主要适用于非极性和弱极性有机物的提取,尚不能对极性(尤其是强极性的)有机成分进行有效定量分析。而极性有机成分对气溶胶颗粒物粒径和理化性质的影响很大,如一元、二元羧酸可作为云凝结核。虽然在线衍生热解吸分析颗粒物中极性化合物的方法已有少量报道,但并未得到广泛应用,原因在于所报道的方法并未得到让人满意的分析结果。因此,极性有机成分分析方法还亟待研究发展。另外,在今后的工作中,仍需对现有的前处理技术不断地改进和完善,发展更加适合于大气气溶胶有机成分分析、高通量、高灵敏、选择性的分析方法。
[1]McMurry P H.Atmos Environ,2000,34(12/13/14):1959
[2]Cassee F R,Roux M E H,Gerlofs N M E,et al.Inhal Toxicol,2013,25(14):802
[3]Turpin B J,Saxena P,Andrews E.Atmos Environ,2000,34(18):2983
[4]Saxena P,Hildemann L.J Atmos Chem,1996,24:57
[5]Hays M D,Lavrich R J.TrAC-Trends Anal Chem,2007,26(2):88
[6]Cochran R E,Dongari N,Jeong H,et al.Anal Chim Acta,2012,740:93
[7]Pietrogrande M C,Bacco D,Chiereghin S.Anal Bio Chem,2013,405(2/3):1095
[8]Jimenez J R,Parshintsev J,Laitinen T,et al.Anal Meth,2011,3(11):2501
[9]CoscollàC,Castillo M,Pastor A,et al.Anal Chim Acta,2011,693(9):72
[10]Hart E,Coscolla C,Pastor A,et al.Atmos Environ,2012,62:118
[11]Schantz M M,McGaw E,Wise S A.Anal Chem,2012,84(19):8222
[12]Piazza R,Gambaro A,Argiriadis E,et al.Anal Bioanal Chem,2013,405(2/3):917
[13]Zhou Y S,Wang H W,Guan Y F.Chinese Journal of Analytical Chemistry(周延生,王涵文,关亚风.分析化学),2004,32(8):983
[14]Zhou Y S,Liu W M,Zhao J H,et al.Chinese Journal of Analytical Chemistry(周延生,刘文民,赵景红,等.分析化学),2005,33(9):1231
[15]Shimmo M,Hyotylainen T,Hartonen K,et al.J Microcolumn Sep,2001,13(5):202
[16]Castells P,Santos F J,Gaiceran M T.J Chromatogr A,2003,1010(2):141
[17]KotianováP,MatisováE,Puxbaum H,et al.Chem Anal,2004,49:833
[18]van Pinxteren D,Teich M,Herrmann H.J Chromatogr A,2012,1267:178
[19]Menezes H C,Cardeal Z D L.J Chromatogr A,2011,1218(21):3300
[20]Alvarez A O,Cuadra R L,Gonzalez I F,et al.Anal Chim Acta,2007,597(2):273
[21]Chow J C,Yu J Z,Watson J G,et al.J Environ Sci Health Part A,2007,42(11):1521
[22]Greaves R C,Barkley R M,Sievers R E.Anal Chem,1985,57:2807
[23]Hays M D,Smith N D,Kinsey J,et al.J Aerosol Sci,2003,34(8):1061
[24]Hays M D,Smith N D,Dong Y J.J Geophy Res Atmos,2004,109(D16):1
[25]Meng H,Zhao J H,Duan C F,et al.Chinese Journal of Analytical Chemistry(孟虎,赵景红,段春凤,等.分析化学),2014,42(7):931
[26]Waterman D,Horsfield B,Leistner F,et al.Anal Chem,2000,72:3563
[27]Gil-Molto J,Varea M,Galindo N,et al.J Chromatogr A,2009,1216(9):1285
[28]Ding L C,Ke F,Wang D K W,et al.Atmos Environ,2009,43(32):4894
[29]Yadav S,Tandon A,Attri A K.J Hazard Mater,2013,252/253:29
[30]Grandesso E,Ballesta P P,Kowalewski K.Talanta,2013,105:101
[31]Falkovich A H,Rudich Y.Environ Sci Tech,2001,35(11):2326
[32]Yu J Z,Huang X H H,Ho S S H,et al.Anal Bioanal Chem,2011,401(10):3125
[33]Ho S S H,Yu J Z,Chow J C,et al.J Chromatogr A,2008,1200(2):217
[34]Ho S S H,Yu J Z.J Chromatogr A,2004,1059(1/2):121
[35]Williams B J,Goldstein A H,Kreisberg N M,et al.Aerosol Sci Tech,2006,40(8):627
[36]Beiner K,Plewka A,Haferkorn S,et al.J Chromatogr A,2009,1216(38):6642
[37]Sheesley R J,Deminter J T,Meiritz M,et al.Environ Sci Tech,2010,44(24):9398
[38]Orasche J,Schnelle K J,Abbaszade G,et al.Atmos Chem Phys,2011,11(17):8977
[39]Jeon S J,Meuzelaar H L C,Sheya S A N,et al.J Air Waste Manage Assoc,2001,51(5):766
[40]Hansen K J,Nansen B N,Cravens E,et al.Anal Chem,1995,67(19):3541
[41]Neususs C,Pelzing M P A,Herrmann H.J Geophys Res,2000,105(D4):4513
