第一性原理研究由金属镍和钇稳定的氧化锆所形成的三相边界微观结构
2014-09-17付召明王明阳张岩星杨宗献
付召明 王明阳 张岩星 张 娜 杨宗献
(河南师范大学物理与电子工程学院,河南新乡453007)
1 Introduction
Solid-oxide fuel cells(SOFCs)1,2have emerged as one of the promising technologies for efficient electrochemical energy conversion because a wide variety of hydrocarbon fuels can be used.The anode is an important constituent part of the SOFC and has been intensively investigated.3-5In recent years,lots of experimental6-8and theoretical studies9-18have focused on understanding the mechanism and kinetics of hydrogen oxidation at the anode of nickel supported on yttria-stabilized zirconia(Ni/YSZ).Although it is well-known that the triple-phase boundary(TPB)between Ni,YSZ,and gas plays a vital role in the SOFCs,the underlying mechanism details are still ambigu-ous and remain a matter of debate.By employing ab initio calculations,it is possible to elucidate the mechanisms of the electronic charge transfer and current generation as a result of the electrochemical oxidation of fuel at the anode.
As shown in the previous works,15-17,19the mechanisms proposed for the electrochemical oxidation of fuel at the anode are directly related to the different Ni/YSZ models constructed by the authors.Therefore,it is important to propose a way to construct a reasonable TPB model of the Ni/YSZ.Generally,for the interface of the oxide and metal,the lattice mismatch brings many difficulties for the density function theory(DFT)modeling.For example,in order to construct the TPB within a small supercell,Shishkin and Ziegler15modeled the Ni/YSZ system with a supported Ni nanowire on the YSZ surface.In this model,although the dimension(length)of the initial Ni nanowire(0.75 nm)is very close to that of the YSZ slab(0.725 nm),the Ni nanowire is destroyed after the geometry optimization,and the(100)surface transforms to the distorted(111)surface with the atom rearrangement.To overcome the defects arising from the lattice mismatch between the Ni nanowire and YSZ substrate,the anode model with an isolated Ni cluster on YSZ is a reasonable choice,in which a bigger Ni cluster and a larger YSZ surface cell have to be adopted in order to obtain a realistic TPB.For the adsorption of a large metal cluster on a large oxide substrate surface,it is conceivable that determining the most stable interfacial structure is an extremely challenging task.Obviously,determination of the stable interfacial geometry requires a search through a large parameter space,spanning the rotational and translational degrees of freedom.This is beyond the capacities of the present DFT calculations,and is especially difficult for the YSZ surface with the Y dopants and intrinsic oxygen vacancies.Therefore,the recent Ni/YSZ models used by Cucinotta16or Ammal17et al.seemingly ignored searching for the most stable interfacial structure.
The electrochemical reaction at the anode of SOFC can be described by the following equation:

In this paper,we combine the classical Monte Carlo(MC)method with DFT calculations to search for various stable adhesion configurations of the Ni46cluster on the YSZ surface adopted in Cucinottaet al.′work,16and evaluate the inaccuracy due to the structural instability.We do find more stable adsorption structures,including new TPBs with the active interface O,which would give the new mechanisms on the electrochemical reactions at the SOFCs.In addition,using the Bader charge analysis that has been adopted in Cucinottaet al.′work,16we systematically investigate the electron transfer in the various Ni/YSZ models(including Cucinottaet al.′model)as removing a surface or bulk O,and explain why the previous DFT simulation can not give the correct number of the transferred charge.
2 Model and computation method
Spin-polarized calculations presented in this work were performed employing the periodic DFT method implemented in the Vienna Ab-Initio Simulation Package(VASP).21The exchange-correlation interactions were treated with the Perdew-Burke-Ernzerhof(PBE)functional.22The electron-ion interactions were treated using the projector augmented wave(PAW)method.23,24The wave functions were expanded in plane waves with a cut off energy of 408 eV.The Monkhorst-Pack k-point mesh of 1×1×1 was used for the Brillouin zone(BZ)sampling.The atoms in the bottom multilayers were kept fixed for all calculations.Structural optimization of all systems was performed until the atomic forces drop below 0.2 eV·nm-1.The Bader charge analysis scheme25was applied to determine the atomic charges and charge transfer.A Ni cluster with 46 atoms(Ni46)was placed on the YSZ surface that consists of 144 O,63 Zr,12 Y atoms,and six constitutional vacancies,which corresponds to 8.7%(molar fraction)of Y2O3in the YSZ model.The YSZ(111)surface was modeled by a(5×5)supercell with a slab of finite thickness(0.75 nm,9 atomic layers)separated by a vacuum layer of 1.5 nm,which is sufficiently large to eliminate slab-slab interactions perpendicular to the surface,and the bottom 3 atomic layers were fixed to mimic the bulk.The positions of the ions in the remaining layers were fully optimized in X,Y,and Z coordinates(under the restriction of fixed cell parameters).Test calculations with a thicker vacuum layer(1.8 nm)showed that the calculated results are actually converged with respect to the vacuum thickness,e.g.,the total energies of adsorption systems(Ni46/YSZ)are almost the same.
For the Ni46/YSZ(111)system,various possible matching patterns at the interface should be taken into account,and each matching pattern corresponds to one adsorption configuration.The classical Monte Carlo(MC)method was used to search for the stable adhesion configurations through the conformational parameter space of the Ni/YSZ adsorption system,spanned by rotating and moving the Ni cluster on the YSZ surface.A set of conformational parameters(θ,ΔX,ΔY,and H)in a four-dimensional sample space were used to depict the different matching patterns,where ΔX(ΔY)was used to depict the relative moving of cluster along X-axis(Y-axis);θ was used to depict the relative rotation of cluster about the Z-axis through the cluster center;H is the distance between two surfaces constructing the interface.For each matching pattern determined by one set of conformational parameters,the number of O―Ni bonds at the interface can be calculated by judging whether the distances of Ni atoms and surface O atoms are close to standard O―Ni bond length.The interfacial O―Ni bond lengths between Ni(111)and YSZ(111)were set to 0.2 nm,26,27and the maximum allowable deviation was set to 0.02 nm in our bonding criterions.In the previous image simulations of high-resolution transmission electron microscopy(HRTEM),28the interlayer distances(dinter)between the Ni and YSZ were set to be 0.195 nm for the O-terminated YSZ(111)model.Here the dintervalues were sampled in the range from 0.185 to 0.205.Thus,we can screen out the Ni/YSZ adsorption configurations that have large number of O―Ni bonds.In this work we considered the configurations with the maximum number of bonds.Significantly,the constitutional vacancies in YSZ adjacent to the surface would have effects on the adsorptions of Ni atoms,so the relative positions between the constitutional vacancies and Ni cluster were taken into account,which can be divided into three cases:the constitutional vacancies lie under the cluster,at the edges of the cluster,and far away from the cluster.By random sampling to the adsorption configurations,the maximum bonding numbers can be calculated according to the mentioned criterions for the three cases,respectively.In this way,we can get a small number of possible stable configurations by omitting the equivalent and similar configurations.Then we performed the DFT optimization to obtain the accurate energies of these systems and get the most stable Ni46/YSZ configuration from them.Fig.1 displays the Ni/YSZ model,with the translational and rotational operations on the Ni46cluster.In this work,we ignored the influence of fuel gas on the TPB structures,and used an ideal model with Ni,YSZ,and vacuum to simulate the actual TPB.
3 Results and discussion
3.1 Energy level

Fig.1 Ni/YSZ model with the translational and rotational operations on the Ni46cluster to go through the conformational space of the Ni/YSZ adsorption system
For the Ni46/YSZ(111)adsorption configuration with Ni46(111)bonded to YSZ(111)surface(shown in Fig.1),we employ a classical MC method to search for the possible stable structures in the special phase space,which are then checked through density functional theory calculations using the VASP.21We find that the Ni/YSZ structure adopted by Cucinotta et al.16is just one of the stable configurations,corresponding to system 1 in Fig.2a.In their work,the adsorption configurations were determined using the DFT method with the PBE exchange correlation functional and norm-conserving pseudopotentials;the Kohn-Sham orbitals were expanded in a triple-zeta valence plus polarization Gaussian-type basis set16.However,more stable configurations(systems 2 and 3)are left out in reference16.Comparedwith the system 1,the most stable configuration(system 3)found by us is lower in energy by 0.7 eV.The relative energies of the three systems are shown in Fig.2b(black line).These results validate our methods in searching for the more stable con-figurations.

Fig.2 Model of the Ni/YSZ triple-phase boundary
3.2 Structure information
The configurations of TPBs have an important influence on the electrochemical reaction for the hydrogen oxidation at the Ni/YSZ anode.The different Ni/YSZ adsorption structures can present different TPBs.So the structural differences of three adsorption systems are presented.The two newly found stable structures(systems 2 and 3)shown in Fig.2(a)have one and two constitutional oxygen vacancies(COVs)close to the vertexes of the Ni46cluster,respectively.This differs from the model used in reference16(system 1),where no COV adjoins the vertexes.We have also tested the single Ni atom adsorption on YSZ surface and found that the most stable adsorption site is just above the intrinsic vacancy site.It might be general that the Ni cluster vertexes favor to locate near the COV.
More importantly,a new type of TPB is formed in the most stable configuration compared with that of the system 1.The TPB in system 1 is shown in Fig.3(a),and the new type of TPB is shown in Fig.3(b).The special interface oxygen atoms are found at the new TPBs,marked by the circle of Fig.3(b).The oxygen atom of this kind only binds to one Zr atom and two Ni atoms with the bond lengths of 0.210 nm(Zr―O)and 0.187 nm(Ni―O),respectively,which is much different from the other interface O atoms.While in the previous work,16,17this kind of special oxygen atom is not observed.In their proposed mechanism,the interface oxygen should overcome a barrier to break one of the two Zr―O bonds and form the oxygen atoms of this type.Interestingly,in our most stable configuration,the active interface oxygen exists directly at the interface.Therefore the active interfacial oxygen in reference16appears as an intermediate states by breaking one of the Zr―O bond intentionally,which is not equivalent to our most stable system because the energy of the latter is lower than that of the former by 1.1 eV.
3.3 Charge transfer
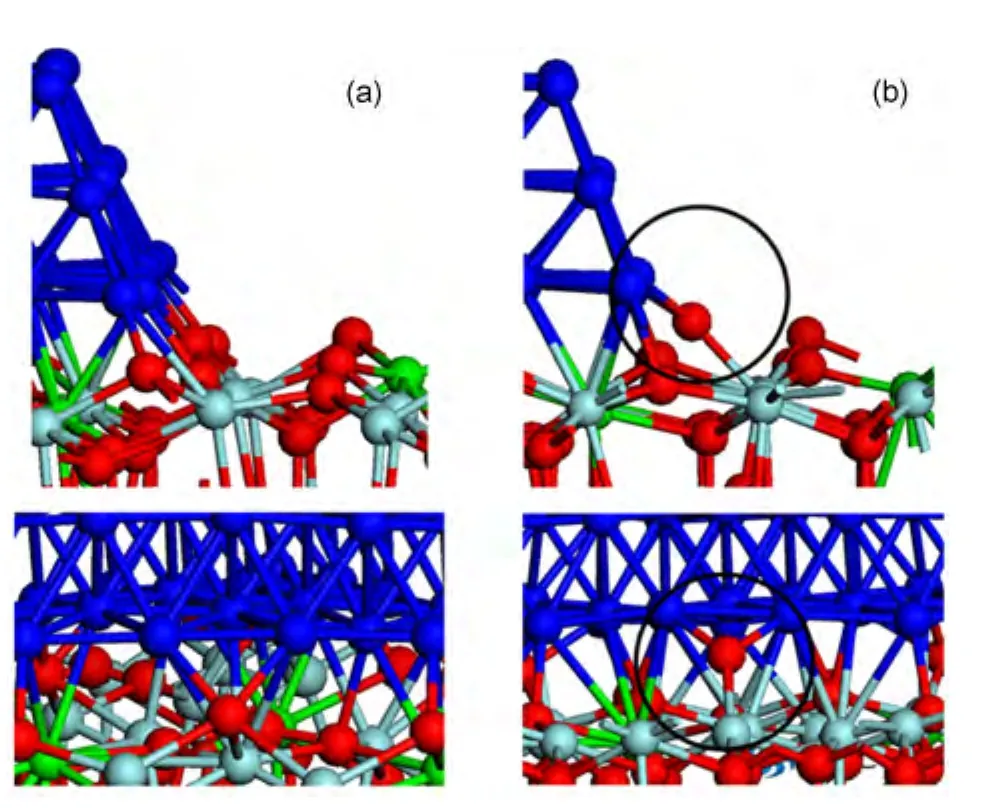
Fig.3 Comparison between the new TPB and the old one
In previous work,researchers found that the adsorbed Ni cluster on YSZ nearly remains electrically neutral.15,26We calculate the charge transfer between the Ni cluster and YSZ for systems 1,2,and 3 by the Bader charge analysis scheme.25It is found that for the more stable systems 2,3,the Ni46clusters only transfer around 0.1e to the substrate,which is in agreement with previous results.15While for the least stable system 1 used in reference,16the Ni46cluster transfers around 0.9e to the substrate.These results indicate that,in the least stable Ni-YSZ configuration,the strong mismatch between the adsorbate Ni46and the substrate YSZ surface results in the excess charge transfer.
In addition,under removing a neutral bulk O atom of YSZ(YSZ-O),the charge transfer from the YSZ to the Ni cluster is critical to understand the mechanism of the electrochemical oxidation of fuel in the anode,and is also regarded as the key point for the simulations of reaction described in Eq.(1).Therefore we focus on three different stable Ni46/YSZ adsorption configurations and calculate the number of electrons transferred(Ntran)with removing a neutral oxygen atom in the YSZ bulk.The Ntranis defined by:

where the NNiis the number of the charge on the Ni cluster.Our main results are summarized in Fig.2b(red dashed line),which suggests that,(1)for different systems the values of Ntranvary in a wide range in responding to the removal of a bulk O;(2)the more stable of a system,the smaller the Ntranis.Particularly,Ntranfor the most stable system is only 0.67e,a value which is in good agreement with some of recent results(0.66).15However,it is far fewer than that in reference16.Furthermore,we have also tested the case with an interface O removed,values of Ntranfor these three systems become 1.0,1.1,and 1.0,which are also in line with Shishkin andZiegler′sresult(1.0)15and Ammal andHeyden′sresult(1.2).17
In fact,as a neutral O is removed from system to form an O vacancy,the distance of the Ni46and this O vacancy(VO)would also affect the charge transfer from YSZ to Ni cluster.Therefore,we investigate the charge transfer in the system 3 for the different Ni-VOdistances.Fig.4 shows that the number of transferred electrons decreases as the distance between the Ni46center and the O vacancy(DV-Ni)increases.Hence the DV-Niis important to exactly describe the charge transfer.It is expected that,with the O vacancy being further away from the Ni part,the fewer electrons would transfer to the Ni part,and the extra electrons under removing O atom would be mainly localized nearby the vacancy.The results will not support the mechanism of electrochemical reaction presented in references.15-17,19
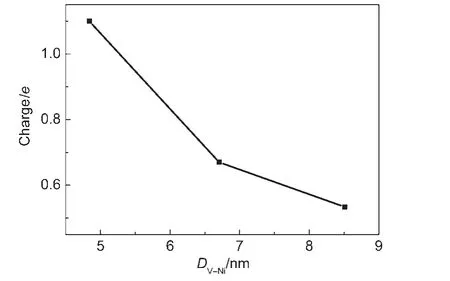
Fig.4 Changes of the transferred charge along with the distance of the Ni46center and the O vacancy for the most stable Ni/YSZ model(system 3)found in this paper
These results suggest that,for a neutral system of the Ni/YSZ with an O vacancy,the Ni/YSZ anode model can not give a rational number of charge transfer,which is also evidenced in the previously published results.15,17,19A possible reason is that the entire electrochemical process depends on the overall system of SOFCs,not just on anodes.And in these theoretical works,only an anode model of Ni/YSZ is simulated.Under the SOFC working conditions,though the whole cell remains electrically neutral,the anode(Ni/YSZ)is negatively charged,and the cathode is positively charged,as shown in Fig.5(a,b).The charge distribution on the anode would be affected by the whole cell system including anode,electrolyte,and cathode.Therefore,it would be inappropriate to use the Ni/(YSZ-O)model to calculate the electron transition in Eq.(1).In addition,the limitations of the conventional DFT to depict electronic exciting should also be taken into account.The sketch in Fig.5 can help to understand the above discussion.
In the Ni/(YSZ-O)model,the extra electrons on Ni are the shared electrons that form the covalent bonds between Ni and reduced YSZ substrate.So they are only localized at the interfacial Ni atoms,as confirmed by the analysis of Bader charges in Fig.6.
The net charge on the interfacial Ni atom layer is 0.64e,which is much close to the transferred charge toward the Ni cluster(0.67e),indicating that the transferred electrons are localized on the interfacial Ni atoms which form covalent bonding with the substrate YSZ-O.In addition,it is also found that the Ni layers away from the interface will remain electrically neutral,as shown in Fig.6.In the case with surface O vacancy,the identical results are given.So these extra electrons will not contribute to the formation of the electromotive force in the SOFCs.According to the foregoing discussion in Fig.4,the shared electrons will disappear as the O vacancy is far away from the Ni cluster.So the charged Ni turns back into the neutral Ni naturally when the O vacancy becomes deeper,which is equivalent to the Ni/YSZ without O vacancy.Just as discussed at the beginning of this part,the Ni is almost neutral in the adsorption system of Ni/YSZ without O vacancy.
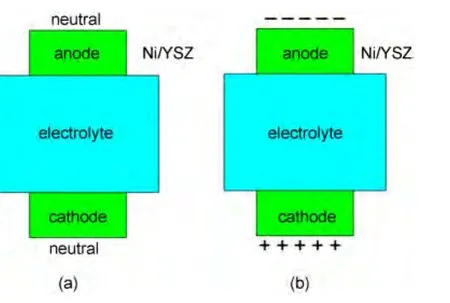
Fig.5 Asketch depicting the neutral and charged electrodes in SOFCs

Fig.6 Distribution of the net charge(Nnet)on each Ni atom along the Z axis upon removing a bulk O from the most stable Ni/YSZ system
In fact,for the metal/oxide systems,it is an extremely common phenomenon that the extra charge is transferred to the metal as an O atom in the oxide is removed,such as the cases in the metal/MgO systems(metal=Cu,Ag,Au),29where there exists a very pronounced transfer of electrons from the vacancy to the metal cluster adsorbates:1.00e,0.99e,and 0.97e on Cu4,Ag4,and Au4,respectively.Therefore this sort of electron transfer is a universal phenomenon of the adsorption systems with the metals on the reduced substrates,which can not be taken as the electrochemical process depicted by Eq.(1).The corresponding transferred number of electrons is of course not 2e.
4 Conclusions
In conclusion,the electrochemical oxidation of fuel is known to occur in a small area close to the anode TPB of Ni,YSZ,and vacuum.16,30The different Ni/YSZ adsorption configu-rations would generate quite different TPBs.Therefore,it is necessary to search for the possible stable adsorption structures in the vast phase space,and investigate the effects of different TPBs on the electrochemical oxidation of fuel.Here,we develop a classical MC method to search for the best way for the modeling of the Ni cluster adhesion on the complex YSZ surface.The most stable configuration presents a new TPB including a special kind of interface oxygen,which was expected to be active to the electrochemical reactions in the previously proposed mechanism.16In addition,the extensive DFT simulations suggest that,for the electrochemical reaction on the Ni/YSZ,it would be inappropriate to use the electronic structure given by DFT to depict the electron transfer based on the Ni/YSZ-O model.Therefore,the detailed mechanism of the electrochemical reaction at Ni/YSZ anode of SOFC should be investigated further.
(1) Ormerod,R.M.Chem.Soc.Rev.2003,32,17.doi:10.1039/b105764m
(2)Williams,M.C.;Strakey,J.P.;Surdoval,W.A.;Wilson,L.C.Solid State Ionics 2006,177,2039.doi:10.1016/j.ssi.2006.02.051
(3)Meng,X.X.;Gong,X.;Yang,N.T.;Tan,X.Y.;Ma,Z.F.Acta Phys.-Chim.Sin.2013,29,1719.[孟秀霞,宫 勋,杨乃涛,谭小耀,马紫峰.物理化学学报,2013,29,1719.]doi:10.3866/PKU.WHXB201305151
(4) Liu,D.D.;Xie,Y.M.;Liu,J.;Wang,J.X.Acta Phys.-Chim.Sin.2014,30,331.[刘丹丹,谢永敏,刘 江,王金霞.物理化学学报,2014,30,331.]doi:10.3866/PKU.WHXB201312241
(5) Lei,Z.;Zhu,Q.S.;Han,M.F.Acta Phys.-Chim.Sin.2010,26,583.[雷 泽,朱庆山,韩敏芳.物理化学学报,2010,26,583.]doi:10.3866/PKU.WHXB20100323
(6)Hansen,K.V.;Norrman,K.;Mogensen,M.J.Am.Chem.Soc.2004,151,A1436.
(7) Sukeshini,A.M.;Habibzadeh,B.;Becker,B.P.;Stoltz,C.A.;Eichhorn,B.W.;Jackson,G.S.J.Am.Chem.Soc.2006,153,A705.
(8) Grgicak,C.M.;Giorgi,J.B.J.Phys.Chem.C 2007,111,15446.doi:10.1021/jp073525n
(9) Bieberle,A.Gauckler,L.Solid State Ionics 2002,146,23.doi:10.1016/S0167-2738(01)01004-9
(10) Bessler,W.G.Solid State Ionics 2005,176,997.doi:10.1016/j.ssi.2005.01.002
(11) Vogler,M.;Bieberle-Hütter,A.;Gauckler,L.;Warnatz,J.;Bessler,W.G.J.Am.Chem.Soc.2009,156,B663.
(12)Goodwin,D.G.;Zhu,H.;Colclasure,A.M.;Kee,R.J.J.Am.Chem.Soc.2009,156,1004.
(13) Anderson,A.B.;Vayner,E.Solid State Ionics 2006,17,1355.
(14) Ingram,D.B.;Linic,S.J.Am.Chem.Soc.2009,156,B1457.
(15) Shishkin,M.;Ziegler,T.J.Phys.Chem.C 2009,113,21667.doi:10.1021/jp905615c
(16) Cucinotta,C.S.;Bernasconi,M.;Parrinello,M.Phys.Rev.Lett.2011,107,206103.doi:10.1103/PhysRevLett.107.206103
(17)Ammal,S.C.;Heyden,A.J.Phys.Chem.Lett.2012,3,2767.doi:10.1021/jz301132b
(18) Rossmeisl,J.;Bessler,W.G.Solid State Ionics 2008,178,1694.doi:10.1016/j.ssi.2007.10.016
(19) Shishkin,M.;Ziegler,T.J.Phys.Chem.C 2010,114,11209.doi:10.1021/jp1030575
(20)Xia,X.;Oldman,R.J.;Catlow,C.R.A.J.Mater.Chem.2012,22,8594.doi:10.1039/c2jm16604f
(21) Kresse,G.;Furthmüller,J.Phys.Rev.B 1996,54,11169.
(22) Perdew,J.P.;Burke,K.;Ernzerhof,M.Phys.Rev.Lett.1996,77,3865.doi:10.1103/PhysRevLett.77.3865
(23) Blöchl,P.E.Phys.Rev.B 1994,50,17953.doi:10.1103/PhysRevB.50.17953
(24) Kresse,G.;Joubert,D.Phys.Rev.B 1999,59,1758.
(25) Henkelman,G.;Arnaldsson,A.;Jónsson,H.Comput.Mater.Sci.2006,36,354.doi:10.1016/j.commatsci.2005.04.010
(26) Christensen,A.;Carter,E.A.J.Chem.Phys.2001,114,5816.doi:10.1063/1.1352079
(27) Jarvis,E.A.;Carter,E.A.J.Am.Ceram.Soc.2003,86,373.
(28)Sasaki,T.;Matsunaga,K.;Ohta,H.;Hosono,H.;Yamamoto,T.;Ikuhara,Y.Mater.Trans.2004,45,2137.doi:10.2320/matertrans.45.2137
(29)Neyman,K.M.;Inntam,C.;Moskaleva,L.V.;Rösch,N.Chem.Eur.J.2007,13,277.doi:10.1002/chem.200600545
(30) Sun,C.;Stimming,U.J.Power Sources 2007,171,247.doi:10.1016/j.jpowsour.2007.06.086
