钯催化交叉偶联法合成7-乙炔基-2-芴基膦酸二乙酯
2014-08-28周舟姚骏毕峰彭亮潘晨晨柳利李法宝
周舟,姚骏,毕峰,彭亮,潘晨晨,柳利,李法宝
(有机化工新材料湖北省协同创新中心(湖北大学),有机功能分子合成与应用教育部重点实验室(湖北大学),湖北 武汉 430062)
钯催化交叉偶联法合成7-乙炔基-2-芴基膦酸二乙酯
周舟,姚骏,毕峰,彭亮,潘晨晨,柳利,李法宝
(有机化工新材料湖北省协同创新中心(湖北大学),有机功能分子合成与应用教育部重点实验室(湖北大学),湖北 武汉 430062)
以芴为原料,通过卤代、Sonogashira偶联、钯催化交叉偶联反应等合成了目标化合物7-乙炔基-2-芴基膦酸二乙酯.采用核磁共振谱(1H、13C、31P NMR)、傅里叶变换红外光谱(FT-IR)、液质联用飞行时间质谱(LC-MS TOF)等对目标化合物进行结构表征,表征数据与化合物结构相符.本文采用一步法直接合成膦酸酯,与传统的两步法相比,操作步骤更加简化,反应条件温和,避免使用五氯化磷、二氧化硫等毒性原料,钯催化交叉偶联反应产率高达91%.
芴;炔;亚膦酸二乙酯;钯催化交叉偶联;合成
芳基膦酸酯是一类重要的具有生理活性的医药有机中间体[1],很多生物活性物质含有磷酸酯结构[2],工业上可用作阻燃剂、聚合物添加剂等.农业方面的研究表明芳基膦酸酯具有较强的杀虫活性[3].刚性、直线型结构的末端炔基可以与过渡金属卤化物反应合成金属炔基配位物,可用于分子导线、发光材料、太阳能电池材料等[4-7].目前合成芳基膦酸酯的方法有: 1) 碘代芳烃与亚磷酸二烷基酯的Na或K盐发生光引发反应[8]; 2) 电化学氧化法合成芳基膦酸酯[9]; 3) 阴离子磷-Fries重排法[10]; 4) 芳基自由基与亚磷酸三烷基酯反应[11]; 5) 过渡金属催化碳-磷键的交叉偶联反应.但这些方法均存在不少需要改进的地方,如使用了价格昂贵、高污染的含膦配体,或者是使用了毒性较大的有机溶剂,以及产率较低等.本文以光学活性的芴为原料,通过卤代、Sonogashira偶联、钯催化交叉偶联反应等合成了目标化合物7-炔基-2-芴基膦酸二乙酯(图1).采用核磁共振谱(1H、13C和31P NMR)、傅里叶变换红外光谱(FT-IR)、液质联用飞行时间质谱(LC-MS TOF)等对目标化合物进行结构表征,确证其结构.

图1 7-乙炔基-2-芴基膦酸二乙酯的合成路线
1 实验部分
1.1仪器与试剂仪器:美国瓦里安AS600(600 MHz)高分辨率核磁共振仪,中国武汉物理与数学研究所WIPM 400(400 MHz)核磁共振仪,Agilent 1260-6224 LC-MS TOF飞行时间液质联用仪、BX FI-IR型傅里叶转换红外光谱仪(美国Perkin-Elmet公司生产,KBr压片)、恒温加热磁力搅拌器、AL-206型电子天平(上海,梅特勒-拖利多公司)、SHZ-Ⅲ型循环水式真空泵(上海荣生化学仪器厂)、DZF-602型真空干燥箱(上海精宏实验设备有限公司)、旋转蒸发仪、磁力搅拌器、氮气保护装置.
试剂:所用试剂均为市售,试剂纯度均为AR.
1.22-溴芴的合成将芴(10.0 g,60 mmol)加入150 mL碳酸丙烯酯中,60 ℃搅拌5 min,观察到固体全部溶解后,快速加入NBS(10.7 g,60 mmol),然后停止加热,室温搅拌4 h后,倒入1 L去离子水,剧烈搅拌3 h,过滤并收集滤渣,干燥后得白色固体12.0 g.产率82%.1H NMR(400 MHz,CDCl3),δ=3.88(s,2H),7.74(s,1H),7.63(t,J=12.0 Hz,2H),7.51(d,J=4.0 Hz,2H),7.35(d,2H).
1.32-溴-7-碘芴的合成将2-溴芴(10.0 g,40.8 mmol),碘(2.6 g,10.3 mmol)和碘酸钠(1.6 g,8.2 mmol)加入150 mL冰醋酸中,边搅拌边缓慢滴加4 mL去离子水和2 mL浓硫酸,加热回流2 h后冷却至室温,过滤,收集滤渣,依次用30 mL冰醋酸、100 mL去离子水洗涤,真空干燥后得淡黄色固体13.1 g.产率87%.1H NMR(400 MHz,CDCl3),δ=3.85(s,2H),7.49(t,J=4.0 Hz,2H),7.60(d,J=4.0 Hz,1H),7.66(s,1H),7.70(d,J=2.0 Hz,1H),7.87(s,1H).
1.42-三甲基硅乙炔基-7-溴芴的合成氮气保护下,将2-溴-7碘芴(10.0 g,27.0 mmol),二氯二(三苯基膦)钯(0.3 g,1.1 mmol),三苯基膦(1.5 g,2.2 mmol),碘化亚铜(0.4 g,2.2 mmol)加入60 mL干燥三乙胺和100 mL二氯甲烷的混合溶液中,冰浴搅拌0.5 h,加入三甲基硅乙炔(3.8 mL,27.0 mmol),0.5 h后撤去冰浴,加热至35 ℃,搅拌8 h.除去溶剂后重新溶于二氯甲烷中,过滤,浓缩得粗品,经柱层析分离(石油醚),得白色固体7.6 g.产率83%.1H NMR(400 MHz,CDCl3),δ=0.26(s,9H),3.86(s,2H),7.50(d,J=4.0 Hz,2H),7.62(t,J=8.0 Hz,2H),7.67(t,J=4.0 Hz,2H).
1.57-三甲基硅乙炔基-2-芴基膦酸二乙酯的合成氮气保护下,将2-三甲基硅乙炔基-7-溴芴(3.0 g,9.0 mmol),四(三苯基膦)钯(0.5 g,0.45 mmol),三苯基膦(1.2 g,4.5 mmol)加入30 mL干燥三乙胺和100 mL甲苯的混合溶液中,5 min后加入亚膦酸二乙酯(1.32 mL,10.4 mmol),搅拌回流6 h,自然冷却至室温,过滤,滤液用水洗涤至中性,分液,有机层用无水硫酸镁干燥后过滤,滤液浓缩后得粗品,经柱层析分离(二氯甲烷∶乙酸乙酯=1∶1,V/V),得淡黄色液体3.0 g.产率84%.1H NMR(400 MHz,CDCl3),δ=0.28(s,9H).1.34(t,J=4.0 Hz,6H),3.91(s,2H),4.04~4.26(m,4H),7.45(d,J=6.0 Hz,1H),7.58(s,1H),7.74(d,J=4.0 Hz,1H),7.83(d,J=4.0 Hz,2H),7.99(d,J=8.0 Hz,1H).
1.67-乙炔基-2-芴基膦酸二乙酯的合成将7-三甲基硅乙炔基-2-芴基膦酸二乙酯(0.70 g,1.76 mmol),K2CO3(0.12 g,0.88 mmol)加入40 mL甲醇中,室温搅拌2 h后过滤,收集滤液,减压浓缩后柱层析分离(二氯甲烷∶乙酸乙酯=4∶1,V/V),得黄棕色油状液体0.52 g.产率91%.IR(KBr,ν/cm-1):3 445,3 287,3 061,2 982,2 929,2 102,1 614,1 462,1 411,1 240,1 023,786.1H NMR(400 MHz,CDCl3),δ=1.35(t,J=6.0 Hz,6H),3.15(s,1H),3.94(s,2H),4.05~4.25(m,4H),7.55(d,J=8.0 Hz,1H),7.70(s,1H),7.77(d,J=8.0 Hz,1H),7.84(d,J=2.0 Hz,1H),7.86(s,1H),8.00(d,J=12.0 Hz,1H).13C NMR(150 MHz,CDCl3),δ=144.87,143.83,143.51,141.04,138.44,131.19,130.74,128.83,128.50,121.51,120.54,120.23,84.01,77.82,62.15(2C),36.65,16.41(2C).31P NMR(240 MHz,CDCl3),δ=15.80,18.54.EI(m/z):327.1(M+,1).
2 结果与讨论
2.1 结构表征



图2 7-乙炔基-2-芴基膦酸二乙酯的IR图
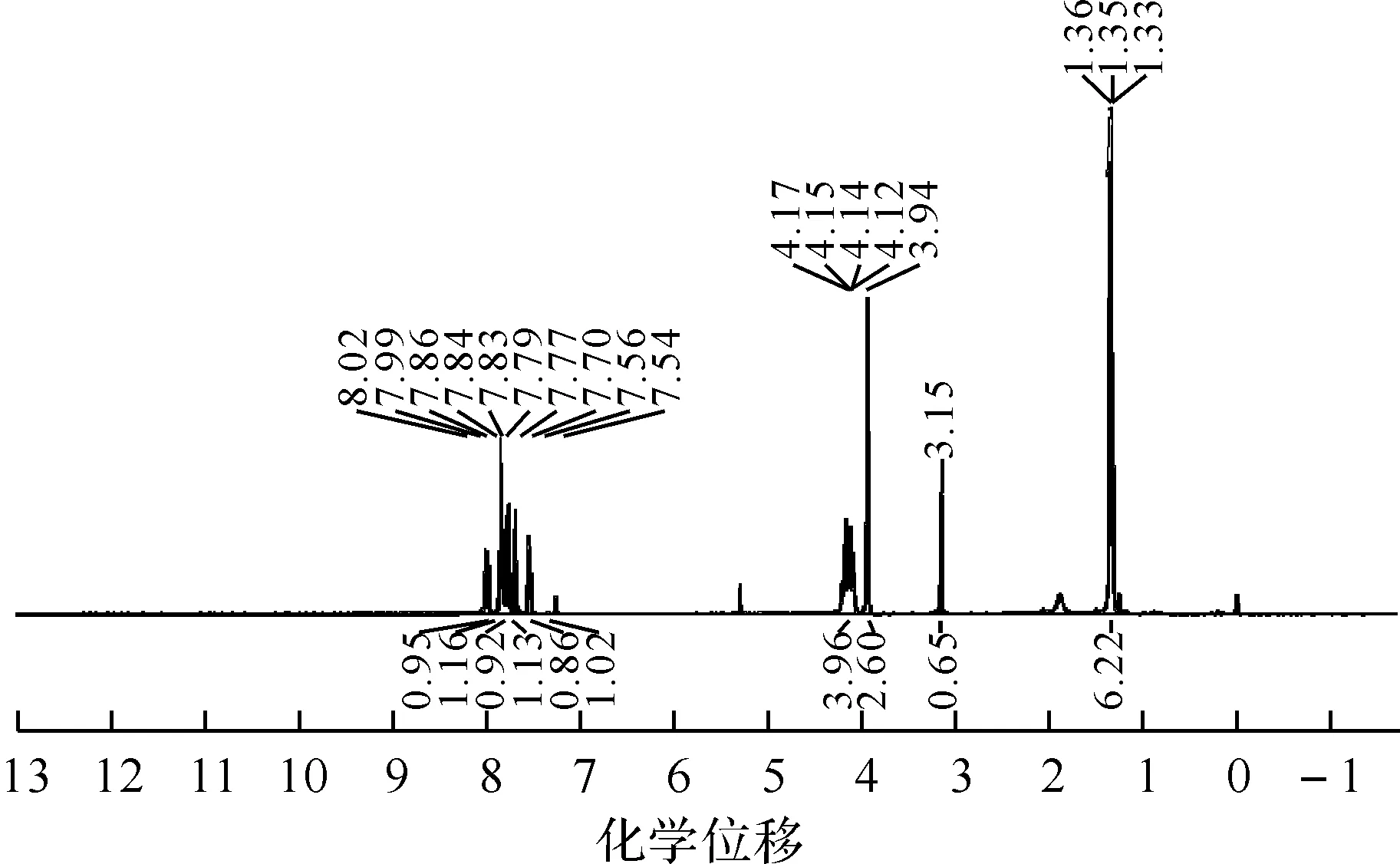
图3 7-乙炔基-2-芴基膦酸二乙酯的1H NMR图

图4 7-乙炔基-2-芴基膦酸二乙酯的13C NMR图


图5 7-乙炔基-2-芴基膦酸二乙酯的31P NMR截图

通过核磁共振谱(1H、13C、31P NMR)、傅里叶变换红外光谱(FT-IR)、液质联用飞行时间质谱(LC-MS TOF)等对7-乙炔基-2-芴基膦酸二乙酯的结构进行了表征,图谱分析结果表明,能够确证该化合物的结构与结构式相符.
2.2目标产物的合成方法分析本文中合成芳基膦酸酯的步骤采用钯催化交叉偶联反应法,反应条件为氮气保护、常压、反应温度为110 ℃,反应时间6 h,整个反应过程及后处理均未使用强酸和强碱,也避免使用强毒性溶剂,钯催化剂用量少,大约为0.01当量,该步反应产率高达91%.由于钯催化交叉偶联反应的反应位点为卤素取代位,因此所得产物选择性高,副产物少,采用柱层析技术分离容易,经柱层析纯化的目标化合物经核磁图、LC-MS-TOF检测,结构获得确证.
目前文献报道的芳基膦酸酯的合成方法,例如:光引发法[8]合成对光照要求严格,对仪器装置要求较高;电化学法[9]合成的反应选择性不高,对含一个取代基的苯环的第二取代位选择性只有33%~40%,其它二取代位副产物难以分离;阴离子重排法[10]合成只对特定的酚羟基芳环结构有效,不具有普遍性;自由基法[11]合成则受到自由基猝灭的因素影响,难以确保产率稳定;常用的氯化磷法[14]合成共分两步,第一步先将芳基化合物与五氯化磷置于冰浴中并通入二氧化硫得到芳基膦酰氯,第二步将膦酰氯转化成膦酸酯,而本文采用钯催化交叉偶联法,采用一步法将膦酸酯基团与芳烃直接偶联,简化了操作步骤,避免使用五氯化磷、二氧化硫等毒性原料,反应条件温和,钯催化交叉偶联反应产率高达91%,是一个绿色、高效的合成方法.
本文中所用合成方法也存在一些不足,如钯催化剂较为昂贵,如果需要大量合成时需要回收催化剂;钯催化交叉偶联反应需要在无氧、氮气保护下进行,需防止催化剂被氧化后导致产率降低.
3 结论
本文中以光学活性的芴为原料,通过卤代、Sonogashira偶联、钯催化交叉偶联反应等合成了未见报道的化合物7-乙炔基-2-芴基膦酸二乙酯.采用核磁共振谱(1H、13C NMR)、傅里叶变换红外光谱(FT-IR)、液质联用飞行时间质谱(LC-MS TOF)等对目标化合物进行了结构表征,确证了结构.与传统的合成法相比,本方法具有反应原料低毒或无毒、反应条件温和、操作简便、产率高,对环境友好等优点.该化合物作为具有良好发光性能的刚性有机配体,可用于合成有机共轭金属配合物,并具有良好的阻燃性及杀虫活性.
[1] Swam Inathan S, Narayanan K V. Rupe and meyer-schuster rearrangements[J]. Chem Rev,1971,71:429-438.
[2] Oish I S, Kang S U, Liu H, et al. Synthesis of A,A-disubstituted 4-phosphonopheny lalanine analogues as conformationally-constrained phosphotyrosyl mimetics[J]. Tetrahedron,2004,60(3):2971-2977.
[3] Ronald K, David C P. Active site generated analogs of reactive intermediates in enzymic reactions: potent inhi-bition of pyruvate dehydrogenase by a phosphonate analog of pyruvate[J]. J Am Chem Soc,1977,99:4504-4506.
[4] Long N J, Williams C K. Metal alkynyl σ-complexes: synthesis and materials[J]. Angew Chem Int Ed,2003,42:2586-2617.
[5] Liu L, Ho C L, Wong W Y, et al. Effect of oligothienyl chain length on tuning the solar cell performance in fluorene-based polyplatinynes[J]. Adv Funct Mater,2008,18:2824-2833.
[6] Wong W Y, Liu L, Shi J X. Triplet emission in soluble mercury(Ⅱ) polyyne polymers[J]. Angew Chem Int Ed,2003,42:4064-4068.
[7] Liu L, Wong W Y, Shi J X, et al. Exploring q-arylcarbazole moiety as the building block for the synthesis of photoluminescent group 10-12 heavy metal diynes and polyynes with high-energy triplet states[J]. Journal of Polymer Science Part A-Polymer Chemistry,2006,44:5588-5607.
[8] Boumekouez A, Jaudet E, Collignon N, et al. A remarkab leaccelerating effect of iodideions in the photo stimulated phosphonylation of bromoaromatic compounds[J]. J Org Chem,1992,440(3):297-301.
[9] Nikitin E V, Romakhin A S, Parakin O V, et al. Electro-chemical synthesis of aryl phosphonates[J]. Russian Chemical Bulletin,1983,32(3):566-568.
[10] Jayasundera K P, Watson A J, Taylor C M. Synthesis of a tetrasubstituted arylphosphonate via the anionic phospho-Fries rearrangement[J]. Tetrahedron Letters,2005,46(25):4311-4313.
[11] Jiao X Y, Bentrude W G. A facile route to vinyl and arylphosphonates by vinyl and aryl radical trapping with (MeO)3P[J]. J Org Chem,2003,68(8):3303-3306.
[12] Informatics division of bio-rad laboratories inc. The sadtler handbook of infrared spactra[M]. Shanghai: Bio-Rad Laboratories Inc,2004:78-79.
[13] Hisanari O, Katsuhiro M, Eiji Y. A helical polyelectrolyte induced by specific interactions with biomolecules in water[J]. J Am Chem Soc,2001,123:7441-7442.
[14] 范望喜,张舟,李文元,等.苯乙烯基膦酸二乙酯阻燃剂的合成与表征[J].化工新型材料,2011,39:71-73.
(责任编辑 胡小洋)
Synthesisofdiethyl7-ethynyl-2-fluorenylphosphonateviaPd-catalyzedcross-couplingmethod
ZHOU Zhou,YAO Jun,BI Feng,PENG Liang,PAN Chenchen,LIU Li,LI Fabao
(Hubei Collaborative Innovation Center for Advanced Organic Chemical Materials(Hubei University), Ministry of Education Key Laboratory for the Synthesis and Application of Organic Functional Molecules(Hubei University), Wuhan 430062, China)
This paper described the synthesis of the title compound diethyl 7-ethynyl-2-fluorenylphosphonate using fluorene as starting material by halogenation, Sonogashira coupling, palladium-catalyzed cross-coupling reactions. The structure of the title compound was characterized and confirmed by NMR (1H,13C and31P), FT-IR, LC-MS TOF spectra. Compared with traditional two steps method, this method has advantages of one-step synthesis of aryl phosphonate, simplification of operation process, avoiding using poisonous PCl5and SO2, moderate reaction condition, and the yield of Pd-catalyzed cross-coupling reaction as high as 91%.
fluorene; alkyne; diethylphosphonate; Pd-catalyzed cross-coupling; synthesis
2013-12-13
国家自然科学基金(21071049)和湖北省自然科学基金(2013CFA087)资助
周舟(1986-),男,硕士生;柳利,通信作者,教授,E-mail:liulihubei@gmail.com
1000-2375(2014)05-0439-04
TQ26
A
10.3969/j.issn.1000-2375.2014.05.010
