ERK1/2/PPARα/SCAD信号途径对生理性和病理性心肌肥大的调控*
2014-08-09黄秋菊黄金贤罗佳妮刘培庆陈少锐潘雪刁臧林泉周四桂
黄秋菊, 黄金贤, 罗佳妮, 刘培庆, 陈少锐, 潘雪刁, 臧林泉, 周四桂△
(1广东药学院临床药学系,广东 广州 510006; 2中山大学药学院药理与毒理学实验室,广东 广州 510006)
心肌是耗能最多的组织之一。哺乳动物胚胎期心脏主要以葡萄糖和乳酸作为能源,出生后则为以脂肪酸氧化为主;但在病理性心肌肥大时脂肪酸氧化降低,糖酵解增加,心肌能量代谢发生“胚胎型转换”[1]。 长期运动训练可使心肌能量代谢水平与适应能力提高,脂肪酸氧化能力增强[2]。深入探讨生理性和病理性心肌肥大2种不同的状态下心肌细胞的能量代谢特征,将有助于进一步认识生理性与病理性心肌肥大的区别。
短链脂酰辅酶A脱氢酶(short-chain acyl-CoA dehydrogenase,SCAD) 是脂酰辅酶A 脱氢酶家族中的一员,特异性地分解短链脂酰辅酶A 底物,是脂肪酸β 氧化的第1个限速步骤,是脂肪酸氧化的关键酶[3]。采用定量蛋白质组学技术我们比较了16周龄自发性高血压大鼠和血压正常大鼠的心肌蛋白质组,首次发现SCAD在自发性高血压大鼠肥大心肌中的表达显著降低[4]。我们进一步证实生理性心肌肥大模型中SCAD mRNA 和蛋白表达均明显上调,而病理性心肌肥大模型中SCAD mRNA和蛋白表达均明显下调,二者之间的表达具有显著差异[5],此外,我们采用体外苯肾上腺素(phenylephrine, PE)刺激诱导的病理性心肌细胞肥大模型中,SCAD在蛋白及mRNA水平上均显著下调,进一步采用RNA干扰技术,观察到SCADsiRNA干扰心肌细胞引起SCAD表达下调的同时,心肌细胞表面积明显增大,心肌肥大标志物心房钠尿因子(atrial natriuretic factor, ANF)和脑钠尿肽(brain natriuretic peptide, BNP)均明显上升[6-7],表明SCAD 表达失调可能与生理性和病理性心肌肥大的发生发展密切相关。
研究表明,心肌肥大发展过程中能量代谢底物的转变与心肌过氧化物酶体增殖物激活受体α(peroxisome proliferator-activated receptor α,PPARα)表达的下降及失活有关[8]。PPARα是核受体超家族成员,也是脂肪酸氧化酶基因的主要转录调控子。研究证实,与野生型小鼠相比,PPARα-/-小鼠心肌中SCAD蛋白表达显著下调,且心肌对脂肪酸摄取、氧化能力下降,表明SCAD基因的表达受PPARα调控[9]。研究报道,PPARα为ERK1/2的下游作用靶点。PE诱导的心肌细胞病理性肥大模型中,激活的ERK1/2使PPARα的表达下调,转录活性下降[10]。然而,研究表明,心脏生理性肥大时ERK1/2并无激活,与病理性心肌肥大的变化趋势不一致[11]。
目前认为,蛋白激酶C激活Raf-1,进而激活有丝分裂原激活蛋白激酶(mitogen-activited protein kinase,MAPK),MAPK信号通路(ERK1/2通路是其中之一)在心肌肥大中发挥着重要作用。然而MAPK信号通路在生理性和病理性心肌肥大中的作用尚未明确。本课题从心肌能量代谢的视角来探讨生理性和病理性心肌肥大的发病机制,揭示生理性和病理性心肌肥大的分子调控机制的不同,丰富对心肌肥大发病机制的认识,以期为预测生理性与病理性心肌肥大及其预后寻找新的分子标志物,并为病理性心肌肥大的治疗寻找新的靶点。
材 料 和 方 法
1 主要试剂和乳鼠心肌细胞培养
RT-PCR测定试剂盒(DRR420A)、TRIzol(9108)和SYBR Green(DRR420S)购于TaKaRa;BCA蛋白定量试剂盒(23225)、Western blotting发光液(34080)购于Thermo;细胞SCAD活性比色法定量检测试剂盒购于上海杰美基因医药科技有限公司;游离脂肪酸测定试剂盒(A042)购于南京建成生物研究所;IGF-1(SRP4121)、PE(P6126)、 单克隆鼠抗PPARα和单克隆鼠抗α-tubulin购于Sigma;单克隆兔抗SCAD购于Abcam;单克隆兔抗p-ERK1/2和单克隆兔抗ERK1/2购于Cell Signaling Technology。
采用本实验室已建立的乳鼠心肌细胞改良法分离并培养心细胞,取2~3 d SD 乳鼠(广州中医药大学实验动物中心) 用胰蛋白酶(Sigma) 冰上消化20 min 后,37 ℃水浴多次消化将乳鼠心脏消化成为单细胞悬液,在差速贴壁分离后,调节细胞密度种于培养皿中,置于37 ℃、5% CO2培养箱中培养,并加入0.1 mmol/L BrdU(Sigma) 抑制成纤维细胞生长。按照上述方法分离制备的心肌细胞,经α-actin 抗体的免疫细胞化学染色,纯度可达95%以上,符合实验要求。实验分为:(1)对照组:正常培养心肌细胞;(2)IGF-1组:加入IGF-1(10 nmol/L)作用24 h;(3)PE组:加入PE(10 μmol/L)作用24 h。
2 方法
2.1心肌细胞表面积的检测 按照实验分组处理细胞,收获细胞后用德国Zeiss(AX10)倒置荧光显微镜200倍摄像,各处理组随机选取3次实验的10个视野,每个视野约包含10个左右心肌细胞,测量100个心肌细胞的面积,以ImageJ图像分析软件分析结果。
2.2Real-time PCR检测mRNA的表达 按照TRIzol(TaKaRa)说明书步骤提取细胞或组织总RNA,采用紫外分光光度计检测RNA样品的260 nm、280 nm波长下的A值,检测纯度并计算出RNA的浓度。参照TaKaRa有限公司RT-PCR试剂盒说明书进行逆转录反应,将反应管放置于PCR仪(Thermo) 中进行逆转录反应。两步法进行PCR扩增反应,按照说明书加入荧光染料(TaKaRa)、序列和RT 产物后在real-time PCR仪(Bio-Rad IQ5)中进行反应。逆转录反应参数: 37 ℃ 15 min,85 ℃ 5 s,4 ℃ (1个循环)。具体引物序列见表1[由生工生物工程(上海)有限公司合成]。

表1 Real-time PCR扩增产物引物序列
2.3Western blotting法检测蛋白表达 提取各组心肌细胞总蛋白,BCA 试剂(Thermo)检测细胞蛋白含量后调整上样量,分装、变性,配置10% SDS 分离胶和5% 浓缩胶进行电泳,电泳结束后转移至PVDF 膜(Bio-Rad),室温封闭1 h 后加入Ⅰ抗(SCAD,1 ∶1 000;PPARα,1∶1 000;p-ERK1/2,1∶1 000;ERK1/2,1∶1 000;α-tubulin,1∶10 000),过夜。漂洗后加入Ⅱ抗,室温孵育1 h,化学发光试剂增强反应,X 线压片曝光、显影、定影,结果采用ImageJ 图像分析系统对条带进行分析。
2.4SCAD活性检测 按照实验分组处理细胞,收获细胞后置于冰上裂解30 min,取上清液用BCA蛋白定量试剂盒定量蛋白。酶活性检测是基于2,6-二氯靛酚钠(2,6-dichlorophenol indophenol, DCPIP)作为人工电子受体,替代黄素腺嘌呤二核苷酸(flavin adenine dinucleotide, FAD),在SCAD的作用下,由短链脂酰辅酶A提供电子,经过硫酸甲酯吩嗪(phenazine methosulphate, PMS)的传递,被还原为无色产物,通过分光光度仪的峰值变化(600 nm 波长),来定量分析SCAD的活性。严格按照说明书采用酶标仪法进行检测。
2.5心肌细胞游离脂肪酸含量测定 游离脂肪酸含量检测原理是游离脂肪酸能与铜离子结合形成脂肪酸的铜盐而溶于氯仿中,其含量与游离脂肪酸含量成正比,用铜试剂测定其中铜离子的含量,即可推算出游离脂肪酸的含量。按照实验分组处理细胞,收获细胞后于冰上用手动匀浆器破碎细胞,取上清BCA蛋白定量试剂盒定量蛋白。严格按照游离脂肪酸测定试剂盒(A042)说明书进行检测,根据吸光率计算游离脂肪酸含量[nmol/(g protein)]。
3 统计学处理
数据以均数±标准差(mean±SD) 表示,采用SPSS 13.0 统计软件处理,组间比较应用单因素方差分析。
结 果
1 各组心肌细胞表面积的变化
由图1可见,与对照组相比,分别用PE和IGF-1刺激心肌细胞后,心肌细胞的表面积均显著增大,表明PE和IGF-1都能诱导心肌细胞产生明显肥大。

Figure 1. Surface area of neonatal rat cardiomyocytes.Mean±SD.n=3.**P<0.01 vs control.
2 各组心肌细胞mRNA表达的变化
由图2可见,与对照组相比,肥大标志物ANF和BNP都明显升高,病理性肥大标志物α-骨骼肌肌动蛋白(α-skeletal actin, α-SkA)在PE刺激下明显升高,而IGF-1刺激无明显变化,与研究报道的结果一致[12-14],这说明PE诱导的病理性肥大模型和IGF-1诱导的生理性肥大模型建立成功。与对照组相比,在PE刺激的心肌细胞中PPARα和SCAD mRNA表达均显著下调,而IGF-1刺激的心肌细胞中PPARα和SCAD mRNA表达均显著上调。PPARα和SCAD mRNA表达在2种不同心肌肥大模型中表现出明显的不一致,表明生理性和病理性心肌肥大的发生发展可能与PPARα和SCAD的表达失调有密切关系。
3 各组心肌细胞蛋白表达的变化
由图3可见,与对照组相比,PE刺激的心肌细胞中PPARα和SCAD蛋白的表达显著下降,IGF-1刺激的心肌细胞中PPARα和SCAD蛋白的表达显著上升,这一变化与PPARα和SCAD mRNA的表达变化一致。然而,与对照组相比,p-ERK1/2蛋白的表达在PE刺激的心肌细胞中显著增加,在IGF-1刺激的心肌细胞中显著下降。这表明PE可能通过刺激ERK1/2磷酸化激活抑制PPARα表达,从而抑制下游SCAD表达,引发病理性心肌肥大的发生;而IGF-1刺激可能通过引起ERK1/2磷酸化受抑制,使PPARα表达上调,从而促进SCAD表达增加引发生理性心肌肥大。
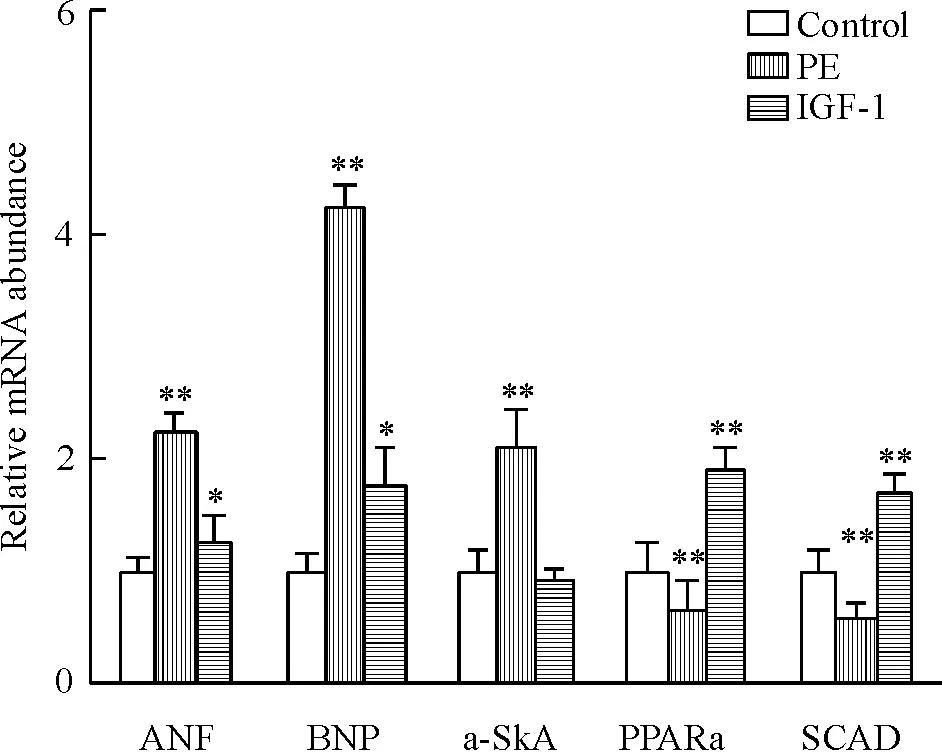
Figure 2. Relative mRNA expression of ANF, BNP, α-SkA, PPARα and SCAD in the myocardial cells.Mean±SD.n=3.*P<0.05, **P<0.01 vs control.

Figure 3. The protein expression of PPARα, SCAD and p-ERK1/2 in the myocardial cells.Mean±SD.n=3.*P<0. 05, **P<0. 01 vs control.
4 各组心肌细胞SCAD的酶活性变化
由图4可见,与对照组相比,PE刺激的心肌细胞SCAD活性显著减弱,而IGF-1刺激的心肌细胞SCAD活性显著增强。各组心肌细胞的SCAD活性与其SCAD mRNA和蛋白表达变化趋势一致。表明在2种不同的心肌肥大中,SCAD不仅在表达量上有不一致的变化,而且酶活性也存在不一致的结果。
5 各组心肌细胞游离脂肪酸含量的变化
由图5可见,与对照组相比,PE刺激的心肌细胞中游离脂肪酸含量显著升高,而IGF-1刺激的心肌细胞中游离脂肪酸含量显著降低。这表明PE刺激的心肌细胞中SCAD蛋白表达和酶活性减低导致了心肌细胞脂肪酸β氧化能力降低,从而使心肌细胞中游离脂肪酸含量增加;而IGF-1刺激的心肌细胞中SCAD蛋白表达和酶活性增加导致了心肌细胞脂肪酸β氧化能力增强,从而使心肌细胞中游离脂肪酸含量下降。
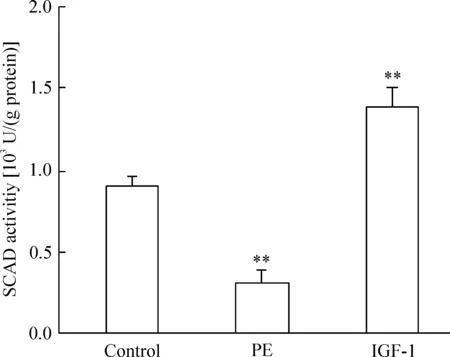
Figure 4. The activity of SCAD in the myocardial cells.Mean±SD.n=3.**P<0.01 vs control.

Figure 5. The content of free fatty acid in the myocardial cells.Mean±SD.n=3.**P<0.01 vs control.
讨 论
运动性心肌肥大本质上是心脏对身体负荷的代偿性机制, 是对运动训练的良好适应性反应,身体训练对于心脏的负荷作用远远小于因持续性和进行性容量或压力负荷过重等病理负荷对心脏的影响[15]。高血压引起的病理性心肌肥大是心脏对急、慢性血流动力学超负荷的一种适应性反应,是心脏为适应长期的压力或容量负荷而产生的代偿性改变,是心力衰竭的前驱病变。越来越多的研究表明,病理性心肌肥大是临床上多种心血管疾病发生率和死亡率增高的一个独立危险因素[16]。因此,探讨生理性与病理性心肌肥大的本质区别,寻找防治病理性心肌肥大的潜在靶点,改善心脏功能,延缓其向心衰发展,对临床心血管疾病治疗有重要意义。
IGF-1作为一种媒介,参与心脏发育、心肌肥厚以及对肌肉体积、力量、身体成分的维持和营养代谢调节的重要作用。越来越多的证据表明,IGF-1在心血管系统中具有特殊作用。它可以促进心肌生长,提高心肌收缩力、心输出量、心室容积并且可以通过刺激收缩和抑制细胞凋亡来维持心脏功能[17]。在诱导心肌细胞肥大的因素中, 神经体液因素是一类常见的原因, 其中肾上腺素受体及其信号转导通路是心肌细胞肥大的一个重要因素。本研究中,IGF-1和PE刺激的心肌细胞表面积与对照组相比均显著增大。同时,在mRNA表达变化研究中,肥大标志物ANF和BNP都明显升高,而病理性肥大标志物α-SkA在PE刺激下升高,而IGF-1刺激无明显变化,与研究报道的结果一致[12-14]。这表明PE诱导的病理性心肌细胞肥大模型和IGF-1诱导的生理性心肌细胞肥大模型都建立成功。
SCAD是脂酰辅酶A 脱氢酶家族中的一员,是脂肪酸氧化的关键酶[3]。本研究中,IGF-1刺激的心肌细胞SCAD mRNA和蛋白表达均显著上调,而PE刺激的心肌细胞SCAD mRNA和蛋白表达均显著下调,两者之间存在显著差异,与蛋白质组学的研究结果一致[4]。这表明SCAD的表达失调可能与生理性和病理性心肌肥大的发生发展密切关系。同时,IGF-1刺激的心肌细胞SCAD活性明显增强,游离脂肪酸含量显著下降,表明IGF-1刺激的心肌细胞中SCAD蛋白表达和酶活性增加可能导致了心肌细胞脂肪酸β氧化能力增强,促进了心肌细胞游离脂肪酸代谢,使心肌细胞中游离脂肪酸含量下降,从而达到心肌细胞能量代谢和功能增强的作用。而PE刺激的心肌细胞SCAD活性明显减弱,游离脂肪酸含量显著增高,表明PE刺激的心肌细胞中SCAD蛋白表达和酶活性下降可能导致了心肌细胞脂肪酸β氧化能力减弱,使得心肌细胞对游离脂肪酸利用减少,使心肌细胞中游离脂肪酸含量增加,这种变化引起了心肌细胞能量代谢紊乱和结构改变以及功能的降低。综上所述,SCAD在2种不同的心肌肥大中具有显著差异,提示SCAD可能成为区别生理性和病理性心肌肥大的分子标志物,并且有可能成为病理性心肌肥大的潜在治疗靶点。
心肌肥大发展过程中能量代谢底物的转变与心肌PPARα表达的下降及失活有关[8]。PPARα是核受体超家族成员,也是脂肪酸氧化酶基因的主要转录调控子。本研究中,IGF-1刺激的心肌细胞PPARα mRNA和蛋白表达均显著上调,而PE刺激的心肌细胞PPARα mRNA和蛋白表达均显著下调,与SCAD mRNA和蛋白表达变化趋势相一致。表明PPARα可能调控SCAD的表达,并且这种表达调控呈正相关性。有研究报道,PPARα为ERK1/2的下游作用靶点。同时,在本研究中,p-ERK1/2蛋白的表达在IGF-1刺激的心肌细胞中显著下降,在PE刺激的心肌细胞中显著增高,而总ERK1/2在各组间无明显变化。这表明ERK1/2可能在磷酸化激活后调控PPARα的表达,并且这种表达调控呈负相关性。
综上所述, IGF-1可能通过刺激心肌细胞中ERK1/2磷酸化受抑制,从而使PPARα表达增加,进一步使SCAD表达和酶活性增加,促使心肌细胞脂肪酸β氧化能力增强,导致生理性心肌肥大;而PE可能通过刺激心肌细胞中ERK1/2磷酸化激活后,从而抑制PPARα表达,进一步使SCAD表达和酶活性降低,导致了心肌细胞脂肪酸β氧化能力减弱,引发病理性心肌肥大的发生。本研究揭示了生理性和病理性心肌肥大的分子机制调控的不同,丰富了对心肌肥大机制的认识。然而,SCAD作为生理性和病理性心肌肥大的分子标志物和病理性心肌肥大的作用靶点的可能仍需要更深入的研究。
[参 考 文 献]
[1] Kolwicz SC Jr, Tian R. Glucose metabolism and cardiac hypertrophy[J]. Cardiovasc Res,2011, 90(2):194-201.
[2] Foryst-Ludwig A, Kreissl MC, Sprang C, et al. Sex differences in physiological cardiac hypertrophy are asso-ciated with exercise-mediated changes in energy substrate availability[J]. Am J Physiol Heart Circ Physiol, 2011, 301(1):H115-H122.
[3] Pena L, Angle B, Burton B, et al. Follow-up of patients with short-chain acyl-CoA dehydrogenase and isobutyryl-CoA dehydrogenase deficiencies identified through newborn screening: one center’s experience[J]. Genet Med, 2012, 14(3):342-347.
[4] Zhou SG, Zhou SF, Huang HQ, et al. Proteomic analysis of hypertrophied myocardial protein patterns in renovascularly hypertensive and spontaneously hypertensive rats[J].J Proteome Res, 2006, 5(11):2901-2908.
[5] 周四桂,王 平,路 遥,等. 短链酰基辅酶A脱氢酶在大鼠生理性和病理性心肌肥大中的作用[J]. 中国病理生理杂志, 2012, 28(11):1921-1927.
[6] 罗佳妮,周四桂,陈少锐,等. 短链脂酰辅酶A脱氢酶与心肌肥大关系的初步探索[J].中国药理学通报, 2013, 29(5):673-642.
[7] 黄金贤,罗佳妮,刘培庆,等. AMPK/PPARα/SCAD信号途径对心肌肥大的调控研究[J]. 中国病理生理杂志, 2014, 30(5):769-778.
[8] Smeets PJ, Teunissen BE, Willemsen PH, et al. Cardiac hypertrophy is enhanced in PPARα-/-mice in response to chronic pressure overload[J]. Cardiovasc Res, 2008, 78(1):79-89.
[9] Watanabe K, Fujii H, Takahashi T, et al. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor alpha associated with age-dependent cardiac toxicity[J]. J Biol Chem, 2000, 275(29):22293-22299.
[10] Meng R, Pei Z, Zhang A, et al. AMPK activation enhances PPARα activity to inhibit cardiac hypertrophy via ERK1/2 MAPK signaling pathway[J]. Arch Biochem Biophys, 2011, 511(1-2):1-7.
[11] Gosslesin H, Béliveau L, Burelle Y, et al. Disparate regulation of signaling proteins after exercise and myocardial infarction[J]. Med Sci Sports Exercise, 2006, 38(3):455-462.
[12] Arantes LA, Aguiar CJ, Amaya MJ, et al. Nuclear inositol 1,4,5-trisphosphate is a necessary and conserved signal for the induction of both pathological and physiological cardiomyocyte hypertrophy[J]. J Mol Cell Cardiol, 2012, 53(4):475-486.
[13] Bhavsar PK, Brand NJ, Felkin LE, et al. Clenbuterol induces cardiac myocyte hypertrophy via paracrine signalling and fibroblast-derived IGF-1[J]. J Cardiovasc Transl Res, 2010, 3(6):688-695.
[14] Kong SW, Bodyak N, Yue P, et al. Genetic expression profiles during physiological and pathological cardiac hypertrophy and heart failure in rats[J]. Physiol Genomics, 2005, 21(1):34-42.
[15] 常 芸. 运动性与病理性心肌肥大[J]. 中国运动医学杂志, 1989, 8(1):35-37.
[16] Takasaki K,Miyata M,Imamura M,et al. Left ventricular dysfunction assessed by cardiac time interval analysis among different geometric patterns in untreated hypertension[J]. Circ J, 2012, 76(6):1409-1414.
[17] Sell C, Baserga R, Rubin R. Insulin-like growth factor 1 (IGF-1) and the IGF-1 receptor prevent etoposide-ieduced apoptosis[J]. Cancer Res, 1995, 55(2):303-306.
