CuBr2/TEMPO/双齿希夫碱体系催化分子氧选择性氧化醇
2014-07-20崔梦佳张月成曹小辉赵继全
刘 柳,崔梦佳,张月成,曹小辉,赵继全
(河北工业大学 化工学院,天津 300130)
CuBr2/TEMPO/双齿希夫碱体系催化分子氧选择性氧化醇
刘 柳,崔梦佳,张月成,曹小辉,赵继全
(河北工业大学 化工学院,天津 300130)
2-吡啶甲醛分别与异丙胺、苯胺及其衍生物缩合制得一系列双齿希夫碱,采用1HNMR 及 FT-IR 等手段对所得产物结构进行表征.以合成的双齿希夫碱为配体与 CuBr2、2,2,6,6-四甲基哌啶-1-氧自由基 (TEMPO) 结合构成催化体系,用于分子氧为氧化剂的醇的选择性氧化反应,考察了该催化体系的催化性能以及希夫碱结构对催化性能的影响.结果表明,所有催化体系在 K2CO3存在下,在V 乙腈 ∶V 水 =2∶1 的混合溶剂中于 50 ℃,可高活性、高选择性地催化分子氧氧化苄基、烯丙基伯醇生成相应的醛.双齿希夫碱配体的结构对催化体系的催化性能影响不大.
2-吡啶甲醛;双齿希夫碱;TEMPO;醇氧化;分子氧
0 引言
将醇选择性氧化为相应的醛或酮是最重要的官能团转化反应之一,在有机合成、药物合成以及大宗化学品生产中占有非常重要的地位[1].传统上这一转化需要化学计量甚至超化学计量的无机氧化剂,如高价态的金属盐、金属氧化物、高价碘等[2-6]来实现.然而,这些方法往往存在选择性低、成本高等缺点,而且反应过程中由氧化剂还原生成的低价态化合物对环境造成严重污染.显然,这些传统方法不符合化工生产绿色化以及经济可持续性发展的要求.因此,采用对环境友好的绿色氧化剂如过氧化氢、分子氧替代传统氧化剂进行醇的氧化非常必要.理论上分子氧是最佳的氧化反应的绿色氧化剂,因为其廉价易得,氧化反应的唯一副产物为水,对环境不造成污染.然而,分子氧是三线态的,与单线态的醇反应是禁阻的,必须经催化剂或催化体系的活化才能用作氧化剂.因此,人们构建了多种用于催化分子氧氧化醇的催化体系[7],尤以基于 2,2,6,6-四甲基哌啶-1-氧自由基(TEMPO)的催化体系受到青睐.TEMPO是可以稳定存在的氮氧自由基,当其与具有氧化还原性质的化合物结合时,在分子氧选择性氧化醇的反应中显示出优良的催化性能.
1984 年,Semmelhack 等[8]首次报道了 CuCl/TEMPO 催化体系,该体系可以在室温条件下于 N,N-二甲基甲酰胺(DMF)中将苄醇、烯丙醇等高效、高选择性地氧化为相应的醛.但该体系的不足之处是催化剂用量较大(底物的 10%),而且对脂肪族醇的氧化往往需要 2 倍化学计量的 CuCl2替代催化量的 CuCl.为克服该体系的缺点,人们进行了广泛研究,发现向反应体系引入双齿螯合氮配体,并辅以碱,反应可在低催化剂投料量下顺利进行[9-15].重要的例子包括 Sheldon 及其同事[9-10]发现的 (bpy)CuBr2/TEMPO 催化体系以及Kumpulainen 和 Koskinen[15]报道的 (bpy)Cu OTf2/TEMPO 体系.二者的共同特点是以 2,2'-联吡啶 (bpy) 为螯合配体,提高铜盐在有机溶剂中的溶解度,同时加入叔丁醇钾或碳酸钾促进反应的顺利进行.Stahl 等[11]则采用 N-甲基咪唑 (NM I) 替代无机碱构建了另一催化体系 (bpy)CuI/TEMPO/NM I,该体系较其他体系反应活性更高,对非活泼性的脂肪醇仍显示较好的催化性能.但这些体系中的螯合配体 2,2'-联吡啶价格昂贵,大规模使用受到限制.因此,采用廉价的氮配体替代 2,2'-联吡啶构建新的醇类氧化反应的催化体系非常必要.Repo[16-17]分别采用二亚胺和吡咯类亚胺替代 2,2'-联吡啶构建催化体系实现了醇的氧化,但反应需在较高温度或较高压力下进行.
我们[18-19]以往的研究发现 2-亚胺基吡啶可作为双齿氮配体与甲基三氧化铼配位改进甲基三氧化铼催化的环氧化反应的催化性能.通过对比 2,2'-联吡啶和 2-亚胺基吡啶的结构发现,二者结构相近,而后者可容易地由 2-吡啶甲醛与伯胺缩合得到.因此,本文拟采用一系列 2-亚胺基吡啶替代 2,2’-联吡啶与CuBr2、TEMPO一起构建新的催化体系用于分子氧为氧化剂的醇的氧化反应,考察该体系对各种结构醇氧化反应的催化性能.
1 实验部分
1.1 实验仪器与试剂
主要仪器:核磁共振波谱由德国BruckerAC-P400 型核磁共振波谱仪测得;红外光谱在德国Brucker公司Vector 22 型傅立叶变换红外光谱仪上测得 (溴化钾压片,4 000 ~ 400 cm1);采用山东鲁南瑞虹化工仪器有限公司SP-6800A型气相色谱仪分析氧化反应产物的组成(PEG20M 气相色谱柱,30m × 0.25mm × 0.50m,FID检测器);采用浙江大学智能信息工程有限公司所生产的N-2000 双通道工作站处理数据.
主要试剂如表1.

表1 主要化学试剂Tab.1 Major chem ical reagents
1.2 双齿希夫碱的合成
1.2.1 双齿希夫碱的合成路线及结构
双齿希夫碱的合成路线及结构如图1所示.
1.2.2 合成方法
2-吡啶甲醛缩-4-硝基苯胺 (s1) 的合成:N2保护下,向 50m L 的三口烧瓶中加入 2.67g(0.02mol)4-硝基苯胺,15m L 无水乙醇,并加入适量活化的 0.4 nm 分子筛.磁力搅拌下滴加 15m L 溶有 2.14 g(0.02mol)2-吡啶甲醛的乙醇溶液,然后加热至回流,反应 5 h.反应结束后冷却,过滤除去分子筛,旋蒸除溶剂,得黄色固体粗品,用苯-石油醚重结晶,真空干燥得s1.收率 83%.1H NMR(400 MHz,DMSO,TMS): = 7.50 ~ 7.52(m,2H, PhH),7.58 ~ 7.61(m,1H,PyH),8.01(dt,J=7.6 Hz,J=1.6 Hz,1H,PyH),8.18(d,J=7.6Hz,1H,PyH),8.29 ~ 8.31(m,2H,PhH),8.61(s,1H,HC=N),8.76~8.78(m,1H,PyH);IR(KBr,cm1) v:3 057,2 919,1 628,1 588,1 506,1 339.

图1 双齿希夫碱的合成路线及结构Fig.1 Synthetic routeof thebi-dentate Schiff basesand their structures
2-吡啶甲醛缩-4-溴苯胺(s2)的合成:以对溴苯胺替代对硝基苯胺,其余步骤同s1 的合成,收率 89%.1H NMR (400MHz,DMSO,TMS): =7.30 ~ 7.33 (m,2H,PhH),7.53 ~ 7.55 (m,1H,PyH),7.61~ 7.65 (m,2H,PhH),7.97 (dt,J=7.6Hz,J=1.2 Hz,1H,PyH) 8.15 (d,J=8.0 Hz,1H,PyH),8.60 (s,1H,HC=N),8.72 ~ 8.74(m,1H,PyH);IR (KBr,cm1) v:3 054,2 995,2 922,1 620,1 566,1 476,826.
2-吡啶甲醛缩苯胺(s3) 的合成:以苯胺替代对硝基苯胺,其余步骤同 s1 的合成,收率 85%.1H NMR(400MHz,DMSO,TMS): =7.28 ~ 7.35(m,3H,PhH),7.43 ~ 7.47(m,2H,PhH),7.52 ~ 7.55(m,1H,PyH),7.97(dt,J=7.2Hz,J=1.2Hz ,1H,PyH),8.16(d,J=8.0Hz,1H,PyH),8.60(s,1H,HC=N),8.72 ~ 8.74 (m,1H,PyH);IR (KBrcm1) v: 3053,2921,1622,1584,1485,781,688.
2-吡啶甲醛缩-4-甲基苯胺(s4)的合成:以对甲苯胺替代对硝基苯胺,其余步骤同s1 的合成,收率 90%.1H NMR (400MHz,DMSO,TMS): =2.34(s,3H,PhCH3),7.23 ~ 7.28(m,4H,PhH),7.51 ~ 7.55(m,1H,PyH),7.95(dt,J=7.6Hz,J=1.6Hz ,1H,PyH),8.14(d,J=7.6Hz,1H,PyH),8.60(s,1H,HC=N),8.70 ~ 8.71(m,1H,PyH);IR(KBr,cm1)v: 3055,2989,2918,2858,1 624,1574,1 502,1 459,1 345,820.
2-吡啶甲醛缩-4-甲氧基苯胺(s5)的合成:以对甲氧基苯胺替代对硝基苯胺,其余步骤同s1 的合成,收率 90%.1H NMR (400MHz,DMSO,TMS): =3.80(s,3H,OCH3),7.02(d,J=8.8Hz,2H,PhH),7.40(d,J=8.4Hz,2H,PhH),7.49 ~ 7.52 (m,1H,PyH),7.94(dt,J=7.2Hz,J=1.6Hz,1H,PyH),8.15(d,J=7.6Hz,1H,PyH),8.63(s,1H,HC=N),8.71(d,J=4.4Hz,1H,PyH);IR(KBr,cm1)v: 3 050,3 002,2 957,2 924,2 833,1 625,1 577,1 504,1 296,1 242,1 035,830.
2-吡啶甲醛缩异丙胺(s6) 的合成:以异丙胺替代对硝基苯胺,最后经减压蒸馏提纯,收率 85%.1H NMR(400MHz,CDCl3,TMS): =1.28(d,J=6.4Hz,6H,2CH3) ,3.61 ~ 3.68(m,1H,CH),7.28~ 7.31(m,1H,PyH),7.73(t,J=7.2 Hz,1H,PyH),7.98(d,J=8.0 Hz,1H,PyH),8.40(s,1H,HC=N),8.64(d,J=4.8Hz,1H,PyH);IR(KBr,cm1)v: 3 058,2969,2862,1647,1589,1466,1 371,1 316.
1.2.3 双齿希夫碱的物理性质
双齿希夫碱的物理性质如表2所示.
1.3 催化醇氧化反应
典型的催化氧化步骤如下:向 10m L两口烧瓶中,加入底物 2mmol,配体s0.04mmol,CuBr2(0.013 4 g,0.06mmol),TEMPO(0.009 4 g,0.06mmol),K2CO3(0.013 7 g ,0.10mmol),CH3CN 2m L,H2O 1m L.加料毕,调节活塞以回流冷凝管顶上氧气球置换体系 3次,然后开启磁力搅拌,升温至 50 ℃进行反应,气相色谱监测反应进程.

表2 双齿希夫碱s1 ~ s6 的物理性质Tab.2 Physicalpropertiesof the bi-dentate Schiff base s1 ~ s6
2 结果与讨论
2.1 催化体系的构建
首先,以苯甲醇的氧化为模型反应,K2CO3为助催化剂,考察了双齿配体 s2 对 CuBr2/TEMPO 催化分子氧氧化醇反应的影响,结果列于表3.由表3 可知,当不加 s2 时,反应 11 h,苯甲醇的转化率只有 25.0%(表3,序号 1);当无TEMPO或 CuBr2存在时苯甲醇几乎不发生反应(表3,序号 2,3);当缺少K2CO3时,转化率只有 32.0%(表3,序号 4).这些结果表明,催化体系中的各组分对苯甲醇氧化反应的顺利进行必不可少;所合成的双齿氮配体s2 象预期的一样可以促进反应的进行.

表3 CuBr2/TEMPO/s2 催化分子氧对苯甲醇的氧化反应Tab.3 Oxidation ofbenzylalcoholw ith O2catalyzed by CuBr2/TEMPO/s2
2.2 催化体系及反应条件的优化
考察了s2 的用量(摩尔分数)对反应的影响,以获得CuBr2/TEMPO/s2 体系的适宜的物料比,结果如表4所示.由表4 可知,随着s2 用量的增加,苯甲醇反应所需的时间逐渐缩短.当s2 用量为 1%时,反应 5 h苯甲醇的转化率为 98.2% (表4,序号 1);当 s2 用量为 2%时,反应 3.5 h苯甲醇氧化完全 (表4,序号 2).s2用量超过 2%后,虽然苯甲醇完全氧化完所需要的时间缩短,但考虑到成本,以s2 的量为 2%为宜.
随后考察了TEMPO的量对反应的影响.如表4 所示,当TEMPO的量为 2%时,反应 6 h苯甲醛的转化率为 98.5% (表4,序号 5);将TEMPO 的量提高到 3%,反应 5h苯甲醇的转化率达到 99.2% (表4,序号 6),继续增加TEMPO的量反应时间进一步缩短到 3.5h,但增至 4%以上时,完成反应所需时间不再变化(表4,序号 2,7).综合考虑,TEMPO的量 3%为宜.
同时,还考察了CuBr2的量对反应的影响,结果亦列于表4.当CuBr2的量为 3%时,反应 6.5h苯甲醛的转化率为 99.1% (表4,序号 9);当减少到 2%时,反应 11.5h,苯甲醇的转化率为 98.0% (表4,序号 8),继续延长反应时间转化率不再变化;若增加CuBr2至 5%,反应 5h苯甲醇反应完全(表4,序号 6),与CuBr2用量 3%时相比略有缩短,所以最后选定CuBr2的量为 3%.
K2CO3用量对反应亦有影响,结果如表4 所示(表4,序号 9-11).随着碱量的增加,苯甲醇完全氧化成苯甲醛所需的时间先缩短后增加,这是因为碱量增加有利于苯甲醇脱质子,促进反应进行,但碱量增加也会消耗部分CuBr2,导致活性降低.由表4 可知,K2CO3的量 5%时与 10%时结果相近,因此,最终将K2CO3的量定为5%.

表4 催化体系中各组分的量对分子氧催化氧化苯甲醇反应的影响Tab.4 Effectsof the loading amountsof the components in the catalytic system on the catalytic oxidation of benzylalcoholw ith O2
最后,考察了温度对反应的影响,结果列于表5.由表5可知,苯甲醇的转化率随着温度的升高而升高.当反应温度高于50℃时,温度的增加对反应的增速缓慢,由 50℃升高到60 ℃,苯甲醇的转化率只增加了 2%.因此,最终选择反应温度50℃.
综合以上实验结果,得到催化体系的优化条件为:配体s2 2%,CuBr23% ,TEMPO 3%和 K2CO35%,反应温度为 50 ℃.
2.3 双齿希夫碱配体结构对醇氧化反应的影响
在优化的反应条件下,分别以苯甲醇、对甲氧基苯甲醇以及对氯苯甲醇为底物,考察了双齿席夫碱配体结构对对催化体系催化性能的影响,结果列于表6.
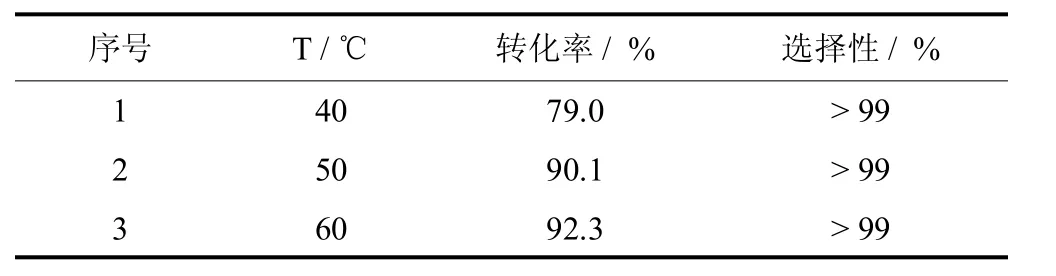
表5 反应温度对分子氧催化氧化苯甲醇反应的影响Tab.5 Effectsof the reaction temperature on the catalytic oxidation of benzylalcoholw ith O2
由表6 可以看出,由 6 个配体构成的催化体系均能在较短时间内高选择性、高活性地将苯甲醇、4-甲氧基苯甲醇和 4-氯苯甲醇转化为相应的醛.其中由芳胺(苯胺或其衍生物)与 2-吡啶甲醛缩合得到的双齿配体s1~ s5 构建的催化体系的催化活性几乎没有差别,即配体苯环上吸电子取代基(-NO2,-Br)或供电子取代基 (-CH3,-OCH3) 对配体构成的催化体系的催化性能无明显影响.但是,由异丙胺双齿希夫碱配体s6构建的催化体系的活性较其他5个稍有下降,可能是异丙胺希夫碱较芳香族伯胺希夫碱空间位阻大(图2B),导致催化循环关键中间体[10,20]存在较大空间位阻,不利于反应的进行.
2.4 底物适用范围的考察
最后在优化的反应条件下,将s1 构成的催化体系应用于包括芳香族和脂肪族等不同结构醇的氧化反应,以考察该催化体系的底物适用性,结果见表7.

图2 催化循环关键中间体中的空间位阻Fig.2 Steric hindrance presentin the key intermediate in the catalytic cycle

表7 s1/CuBr2/TEMPO/K2CO3体系催化分子氧对不同醇的氧化Tab.7 Oxidation of variousalcoholsw ith O2catalyzed by s1/CuBr2/TEMPO/K2CO3
由表7可知,由双齿希夫碱构建的催化体系对芳伯醇、烯丙基伯醇的氧化有很好的催化性能,苯甲醇及单取代的苯甲醇都可以在一定时间内几乎定量地转化为相应的醛(表7,序号 1 ~ 5);烯丙基伯醇中的肉桂醇和香叶醇分别反应 3.5 h 和 5.5 h 就可转化为相应的醛,转化率和选择性都接近 100%(表7,序号 6,7).然而,该催化体系对 1-苯乙醇的氧化几乎没有活性,反应 5h只检测到 1%的苯乙酮生成(表7,序号 9).原因是 1-苯乙醇分子上甲基的空间位阻抑制了醇与铜的配位,不能使TEMPO夺取底物醇的-H[9-10].对于芳杂醇糠醇(表7,序号 8) 和 2-吡啶甲醇 (表7,序号 10),反应 4.5 h的转化率分别只有 10%和 11%,可能原因是底物中的杂原子与 Cu2+配位[21],使催化体系失去活性所致.脂肪族伯醇正辛醇则完全没有被氧化(表7,序号 11),可能原因是脂肪族伯醇羟基上质子不活泼,K2CO3不能将底物转化为醇负离子,进而无法与 Cu2+配位,脱氢反应不进行[9-10].这些结果证明由双齿希夫碱构建的催化体系与 2,2'-联吡啶为配体构建的催化体系性能基本一致.
3 结论
由 2-吡啶甲醛与不同结构的伯胺缩合制得双齿希夫碱配体,这些配体与 CuBr2/TEMPO 构建的催化体系在K2CO3存在下可催化分子氧将苄基及烯丙基伯醇高活性、高选择性地氧化为相应的醛.这些双齿希夫碱配体与 2,2'-联吡啶在促进醇的选择性氧化方面性能相近,但制备方法简单,原料价廉易得,可较大规模使用.
[1]Tojo G,Fernández M.Oxidation of alcohols to aldehydesand ketones: aguide to currentcommon practice[M].New York: Springer,2006.
[2]March J.Advanced organic chem istry: Reactions,mechanisms,and structure[M].4th ed.New York: JohnWiley&Sons,1992.
[3]M ijsW J,Jong D,EdsC.Organic synthesesby oxidationwithmetal compounds[M].New York: Plenum Press Inc,1986: 423-443.
[4]DessD B,Martin JC.Readily accessible12-I-51oxidant for the conversion of primary and secondary alcohols to aldehydesand ketones[J].JOrg Chem,1983,48:4155-4156.
[5]Frigerio M,Santagostino M,Sputore S,etal.Oxidation of alcoholsw ith o-iodoxybenzoic acid (IBX) in DMSO: a new insight into an old hypervalent iodine reagent[J].JOrg Chem,1995,60: 7272-7276.
[6]More JD,Finney N S.A simpleand advantageousprotocol for theoxidation of alcoholsw ith o-iodoxybenzoic acid (IBX)[J].Org Lett,2002,4(17):3001-3003.
[7]Zhan B Z,Thompson A.Recentdevelopments in theaerobic oxidation of alcohols[J].Tetrahedron,2004,60: 2917-2935.
[8]Semmelhack M F,Schm id C R,CortésD A,etal.Oxidation of alcohols to aldehydesw ith oxygen and cupric ion,mediated by nitrosonium ion [J].JAm Chem Soc,1984,106,3374-3376.
[9]Gamez P,Arends IW C E,Reedijk J,et al.Copper(II)-catalysed aerobic oxidation of primary alcohols to aldehydes[J].Chem Commun,2003(19): 2414-2415.
[10]Gamez P,Arends IW C E,Sheldon R A,etal.Room temperatureaerobic copper-catalysed selectiveoxidation of primary alcohols to aldehydes [J].Adv Synth Catal,2004,346(7): 805-811.
[11]Hoover JM,StahlSS.Highly practicalcopper(I)/TEMPO catalystsystem for chemoselectiveaerobic oxidation of primary alcohols[J].JAm Chem Soc,2011,133(42): 16901-16910.
[12]Hoover JM,Ryland B L,StahlSS.Mechanism of copper(I)/TEMPO-catalyzed aerobic alcoholoxidation[J].JAm Chem Soc,2013,135(6): 2357-2367.
[13]Velusamy S,Srinivasan A,Punniyamurthy T.Copper(II) catalyzed selectiveoxidationofprimaryalcohols toaldehydesw ithatmosphericoxygen [J].Tetrahedron Lett,2006,47(6): 923-926.
[14]Mannam S,AlamsettiSK,SekarG.Aerobic,chemoselectiveoxidationofalcohols to carbonylcompoundscatalyzed by aDABCO-Coppercomplex underm ild conditions[J].Adv Synth Catal,2007,349(14-15): 2253-2258.
[15]Kumpulainen ET T,Koskinen AM P.Catalytic activity dependency on catalystcomponents in aerobic copper-TEMPO oxidation[J].Chem Eur J,2009,15(41): 10901-10911.
[16]Figiel P J,Leskela,Repo T.TEMPO-copper(II) diimine-catalysed oxidation of benzylic alcohols in aqueousmedia[J].Adv Synth Catal,2007,349:1173-1179.
[17]FigielPJ,Sibaouih A,Ahmad JU,etal.Aerobicoxidationofbenzylicalcoholsinwaterby 2,2,6,6-tetramethylpiperidine-1-oxyl(TEMPO)/ copper(II)/2-N-arylpyrrolecarbaldim ino complexes[J].Adv Synth Catal,2009,351:2625-2632.
[18] 高宇, 张月成,赵继全.甲基三氧化铼席夫碱配合物的合成与晶体结构 [J].无机化学学报,2009,25(9):1686-1689.
[19] 高宇,张月成,赵继全.双氮席夫碱配体对甲基三氧化铼催化烯烃环氧化反应的影响 [J].催化学报,2009,30(12):1243-1247.
[20]Dijksman A,Arends IW CE,Sheldon RA.Cu(Ⅱ)-nitroxyl radicalsascatalytic galactoseoxidasemimics[J].Org BiomolChem,2003,1(18):3232-3237.
[21]Hara T,Sawada J,Nakamura Y,etal.An anionic D-valine-palladium(Ⅱ)complex supported on ahydroxyldouble saltwith a Br nsted basic phosphateanion:application foraheterogeneous catalyst toward aerobic alcoholoxidation[J].Catal SciTechnol,2011,1:1376-1382.
[责任编辑 田 丰]
Oxidation of alcoholsw ithmolecularoxygen catalyzed by CuBr2/ TEMPO/bidentate Schiffbase ligand catalytic system
LIU Liu,CUIMeng-jia,ZHANG Yue-cheng,CAO Xiao-hui,ZHAO Ji-quan
(Schoolof Chemical Engineering,HebeiUniversity of Technology,Tianjin 300130,China)
A seriesof bi-dentate Schiff base ligandswere respectively synthesized by the condensation of 2-pyridinecarboxaldehydew ith isopropylam ine,aniline and its derivatives.Their structureswere characterized by1H NMR and FTIR.Catalytic systemswere established w ith the synthesized bi-dentate Schiffbasesas ligands in combinationw ith CuBr2and 2,2,6,6-tetramethylpiperidinyl-1-oxy(TEMPO).The catalytic systemswereapplied to theoxidationofalcoholsw ith O2asan oxidant.Theeffectof the structuresof the ligandson the performancesof the catalytic systemswasinvestigated. The results indicate that the catalytic systems can catalyzemolecularoxygen oxidizing primary benzyland allylalcohols to their corresponding aldehydesw ith high activity and selectivity at50 ℃ in CH3CN/H2O(V/V=2/1).The structuresof the bi-dentate Schiff baseshaveno obvious influence on the performance of the catalytic systems.
2-pyridinecarboxaldehyde;bi-dentate Schiff base;TEMPO;oxidation of alcohols;molecularoxygen
1007-2373(2014)05-0042-07
O641.4
A
10.14081/j.cnki.hgdxb.2014.05.008
2014-07-08
国家自然科学基金(21276061);河北省自然科学基金(B2013202158);高等学校博士学科点专项科研基金(20121317110010)
刘柳(1989-),女(汉族),硕士生.通讯作者:赵继全(1963-), 男(汉族),教授,E-mail:zhaojq@hebut.edu.cn .
