Meta-Mesh
——元基因组数据分析系统
2014-06-09苏晓泉宋宝兴王雪涛马新乐徐健宁康
苏晓泉,宋宝兴,王雪涛,马新乐,徐健,宁康
中国科学院青岛生物能源与过程研究所,山东 青岛 266101
生物信息学与计算生物学
Meta-Mesh
——元基因组数据分析系统
苏晓泉,宋宝兴,王雪涛,马新乐,徐健,宁康
中国科学院青岛生物能源与过程研究所,山东 青岛 266101
苏晓泉, 宋宝兴, 王雪涛, 等. Meta-Mesh——元基因组数据分析系统. 生物工程学报, 2014, 30(1): 6−17.
Su XQ, Song BX, Wang XT, et al. Meta-Mesh: metagenomic data analysis system. Chin J Biotech, 2014, 30(1): 6−17.
随着元基因组数据的不断增多,建立一个包含高品质的元基因组样本 (也称为“微生物群落”) 数据的集成化的分析平台成为可能,使得微生物群落样本能够被有效分析、比较与搜索,从中发现更加深入的生物学意义。然而,一方面目前大部分元基因组数据库仅仅提供了简单的数据存储,缺乏良好的样本注释或者仅仅提供了很少的分析功能。另一方面,用于计算微生物群落数据相似性的方法所能够接受的样本数据量非常有限。长期以来,科学家们一直在寻找有效的方法计算海量微生物群落之间的相似性,从而研究样本之间的相似度并发现元基因组数据信息的相关性。Meta-Mesh是一个全新的在线元基因组分析系统,它包括元基因组数据库和分析平台,可以对元基因组样本进行系统、有效地分析,并实现样本的群落结构比较和精确搜索。其中,元基因组数据库已经从公共领域和内部实验室收集了超过7 000个高品质、带有有效注释的样本。同时,Meta-Mesh的分析平台提供了多种在线分析工具,可以对元基因组样本进行群落的结构分析与注释,多角度比较,并能通过快速索引策略和群落结构相似性算法在数据库中高效搜索近似的样本。Meta-Mesh通过“人体微生物群落样本的数据库搜索识别”以及“基于相似度矩阵的样本的聚类”等一系列的元基因组研究案例证明了其分析方面的性能。作为一个在线的元基因组数据库和分析系统,Meta-Mesh将服务于元基因组样本的快速分析、识别、比对、搜索等相关领域。
微生物群落,元基因组,数据库,数据挖掘,在线服务,相似性网络
1 研究背景
众所周知,微生物在地球上无处不在,且数量巨大[1]。它们通常以群落的方式生存,每个群落有不同的结构。因此,微生物群落拥有最大的基因库和遗传功能信息,并应用在大量生物相关的学科,包括生物医药、生物能源、生物修复和生物防御等[2]。
由于超过90%的菌株在微生物群落中不能被孤立或培养[3],元基因组方法已被用来分析一个微生物的群落。这能够从基础的基因层次上进行微生物、群落、栖息环境关系的探索。了解微生物群落内物种多样性 (α多样性) 和微生物群落之间的差异 (β多样性) 已经成为元基因组学研究中的两个最重要的问题[4-5],其中理解β多样性对于研究微生物生态学尤其关键。例如,人类微生物群项目[6]以及相关的人体各个位置的微生物群落研究,包括皮肤[7-8]、肠道[7]、口腔系统[9-10]等,已显示出惊人的多样性。此外,即使来自相似环境的微生物群落也可能有很大的不同[11]。
然而,由于元基因组数据本身的特性,如群落结构的不均衡、数据来源的多样性、尚不成熟的质控标准、数据的极端高通量等,元基因组数据的生物信息学分析往往成为有效设计与解析相关微生物群落的核心瓶颈之一。另一方面,新一代测序技术已经能够进行大量元基因组样本的快速分析。随着公共信息库和世界各地的实验室提供的微生物群落元基因组样本数量迅速增加,对比不同的微生物群落以及大规模的搜索相似样本越来越重要。
1.1 微生物群落元基因组研究的重要性
由于群落中的大多数微生物尚不可培养,直接鉴定群落的元基因组是目前最重要、最迅速的菌群结构与功能的认识方法之一。目前常用的办法是使用细菌、古菌和真菌特异性引物进行系统发育标记分子 (Phylogenetic marker,如16S rRNA) 的扩增,并通过测定其序列来识别微生物群落的物种组分并定量其相对丰度。微生物群落的结构信息也可以通过对群落的所有DNA进行元基因组测序而得到。全长DNA测序和16S rRNA这两种技术手段相结合,不仅可以获得微生物群落中物种组分的信息,而且还能够在基因及其功能水平上对群落中的微生物进行解析和比较。正是基于上述这两种技术手段的结合,使得我们对一些重要的产能微生物群落的结构和功能的认识迅速取得了重大突破[12]。元基因组已成为挖掘与认识自然界高效分解生物质的“生物能源菌群”的结构、功能及其运作机理的代表手段之一[13-14],同时也渗透到环境生物监测与治理[15-16]、极端环境[17]、营养与健康[18-19]等以利用或克服复杂微生物群落及其产物为目的的科学领域。
1.2 微生物群落分析和对比
目前主要有两种方法用于比较和聚类不同的微生物群落样本。第一种是以群落物种的生物分类结构 (Taxonomy) 为基础的方法,首先分析样本的生物分类学结构,然后互相比较。最近的几项焦磷酸测序的研究已经基于这种方法开发。MEGAN[9]是一个进行元基因组的比较[10]和统计分析[20]的工具,它只能进行元基因组样本的两两比较,STAMP[21]也是如此。诸如MG-RAST[8]、ShotgunFunctionalizeR[20]、Mothur[22]、MetaRep[23]等其他方法都使用标准的统计测试 (主要是经过一些修改的t-检验)识别样本之间的差异。然而,这些方法中并不能很好地适用于元基因组数据,从而导致片面的结果[24]。第二种是基于群落物种进化关系(Phylogenetic) 的方法,例如UniFrac[25]和Fast UniFrac[26],是利用样本进化树的重叠部分进行距离计算[27]。然而,这些方法大多是基于复杂的计算[25],难以在考虑效率和精度的同时进行海量样本的扩展:具体来说,当样本数超过数千时这些方法比较或搜索元基因组样本的效率是低下的。
1.3 微生物群落数据库
由于微生物群落样本生成的速度越来越快,一个提供元基因组数据存储,组织以及数据分析和挖掘等功能的分析存储系统对世界范围的多学科用户群有重大价值。然而,目前的元基因组数据库,如MG-RAST[8]和CAMERA2[20]等,主要提供数据存储库的服务,没有提供工具进行基于phylogenetic的样本的比较分析与大范围的搜索能力。新的MeganDB数据库 (http://www.megandb.org/megan-db/home/)对归档的公共元基因组样本进行适当的前处理,可以有效地对存储的样本进行比较以查询样本。尽管如此,MeganDB基于群落物种生物分类学比较方法仍然有局限性。另一方面,本文作者最近开发了元基因组数据库的可扩展的索引和搜索引擎Meta-Storms[28],但缺乏一个组织良好的元基因组数据库,未形成高效的集成化系统。
1.4 高效的元基因组数据库分析和搜索平台
为了进行大规模的元基因组样本的比较分析和搜索,关键是设计一个高效的分析系统和搜索方法,以及精心挑选的元基因组注释数据库。这样一个系统的主要特点包括:1) 一个存储有高质量样本的元基因组数据库及其注释;2) 集成高性能元基因组数据分析和可视化的工具;3) 用于不同的微生物群落之间比较及样本搜索的元基因组搜索计算引擎。
本文将介绍一个集成的微生物群落分析系统Meta-Mesh,它包括一个组织严密的元基因组数据库,一套高效的分析工具,以及易于使用的基于Web的用户界面。Meta-Mesh数据库的总体方案如图1所示。在这个数据库中,各种资源的元基因组数据集被自动收集,手工筛选,并在预处理后注释其群落物种的生物分类学结构。同时,Meta-Mesh生成一个便于进行数据库快速查找的索引。Meta-Mesh的“工作中心(Work Center)”提供相关的分析功能,如数据库浏览,群落结构分析,数据比较,数据库检索等。Work Center支持自定义配置分析流程,并提供数据交互和分享。“用户管理(User Management)”模块能够确保用户的数据隐私和安全。
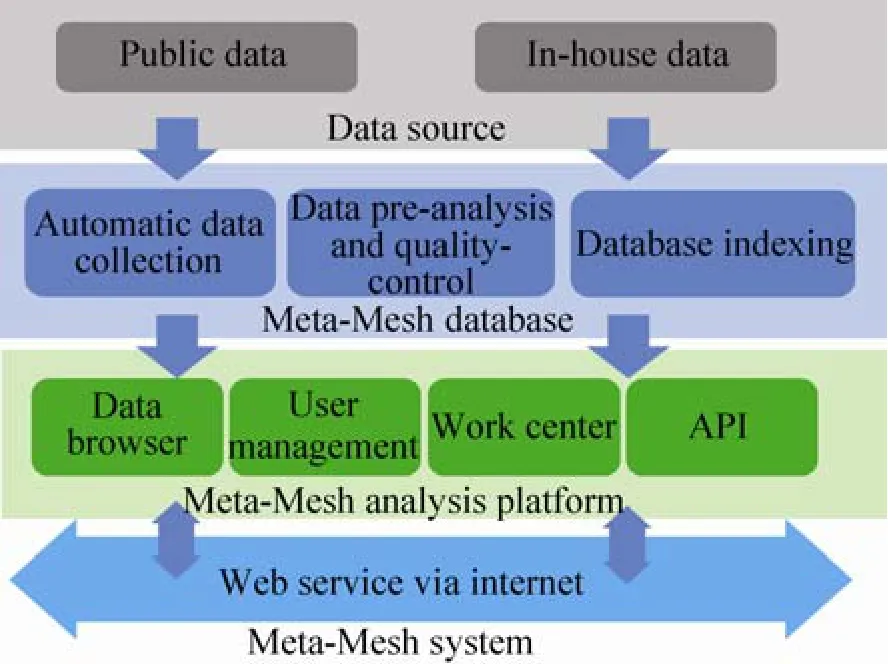
图1 Meta-Mesh系统的总体方案Fig. 1 Overall structure of Meta-Mesh system.
2 研究方法
Meta-Mesh是一个用于元基因组在线分析的高效集成化系统,它为微生物群落研究提供了群落结构分析、数据可视化、样本比较、数据库检索和元基因组数据挖掘的功能。Meta-Mesh网址是http://www.meta-mesh.org/。
在Meta-Mesh系统中,数据、功能和其他资源被组织成4层,都可以通过Web服务进行访问,并提供一个用户友好的用户界面 (图2)。在“数据源(Data Source)”层,Meta-Mesh集成了来源广泛的数据,并组织到一个有良好注释的数据库中。其中所有的数据和资源,可以自由访问和下载。在“工作中心(Work Center)”层,Meta-Mesh提供了一个多功能的分析平台,包括样本结构分析、比较、搜索以及大规模数据挖掘。此外,Meta-Mesh有完整的用户管理策略,允许用户安全高效地上传和分析自己的数据,以及与他人共享数据。在“设施(Facility)”层,基于Web的API (Application programming interfaces,应用编程接口) 的插件和扩展功能允许开发人员进行开发。关于系统的所有文件存储在“文档(Document)”层。
2.1 数据库
2.1.1 自动化的数据搜集和注释
Meta-mesh从大量的公开数据库收集微生物群落资源信息,包括MG-RAST[8]、CAMERA2[20]、HMP (http://www.hmpdacc-resources.org)、GenBank(http://www.ncbi.nlm.nih.gov/genbank/)和我们内部的数据库Q I B E B T-C A S (http://www.computationalbioenergy.org)。为了实现这一目标,本文设计了一个自动数据收集器BEGC-SPIDER (http://autoupdate.meta-mesh.org/)用于访问公开可用的元基因组相关的项目和论文,并将这些原始数据搜集到Meta-mesh数据库中的缓冲区中。
Meta-Mesh保留了所收集数据的原有的信息 (元数据,meta-data),并提供元基因组样本的详细信息,包括项目名称、样本收集地点、日期和时间、源标签和测序方法。对于部分甚至全部元数据缺失的样本也可实行手动标注。基于自动采集和人工筛选,Meta-Mesh在数据库中已经收集了超过12 000个微生物群落样本的元基因组序列数据,并积累了来自209个不同的项目的约7 000个高品质注释样本。此外,BEGC-SPIDER每周更新,使Meta-Mesh数据库中所有数据集定期更新。

图2 Meta-Mesh的Web服务界面设计Fig. 2 Web service portal design of Meta-Mesh.
2.1.2 元基因组数据的质量控制和预处理
首先原始数据缓冲区的样本由QC-Chain[29]进行质量控制评估,然后利用Parallel-META微生物群落分析软件 (由本文作者开发)[30]对数据进行预处理从而进行群落结构和物种进化结构的分析。Parallel-META使用HMM算法(Hidden Markov Model)[31]提取16S rRNA和18S rRNA的生物标记基因片段,然后将其映射到GreenGenes[32]、RDP[33]、Sliva[34]以及Oral Core[35]数据库中进行物种识别、注释、进化分析。在数据分析中,含有300个以上生物标记序列 (16S rRNA或18S rRNA基因) ,且数据库 (GreenGnes,RDP,Sliva,或Oral Core)序列映射率大于等于75%的元基因组数据才能添加到数据库中,以保证Meta-Mesh数据库的高质量。
2.1.3 数据库管理和索引
Meta-mesh数据库中经过质量控制和预处理后的样本,由Meta-Storms[28]数据库引擎进行维护,并提供高效的搜索索引策略。Meta-Mesh通过索引将数据库来划分成更小的子数据集,每子数据集包含结构相似的样本,并将它们分配到适当的位置索引以便快速查找。通过这种方式Meta-Mesh不需要在数据库中搜索所有样本,而是仅仅查询对应于索引的子数据集。因此,搜索速度被显著提高,且无明显的精度损失 (Meta-Storms由本文作者开发,详细搜索算法请参阅文献[28])。
2.2 分析平台
2.2.1 元基因组数据浏览
Meta-Mesh中的元基因组数据以项目为单位进行组织,通过web所提供的界面可以对项目以及其中的样本数据进行浏览,并支持项目的名称的关键字过滤 (图3A)。在每个项目中,元数据、注释、可视化的结构分析结果和样本数据库中的所有相关信息可以通过元基因组样本列表访问 (图3B)。在Meta-Mesh数据库中,用户能够通过高效的Meta-Storms[28]索引机制快速地获得与待查询样本结构相似的其他样本。
2.2.2 用户管理和数据分享
为了确保数据的保密性和安全性,Meta-Mesh只对注册用户 (可通过web注册,登录信息参考附件3,部分具体数据作为附件,包括附件1−3和附表S1−S6,可在网络版中下载)提供数据的上传和处理服务。用户的数据和信息都保存在系统中,并设置为私有数据,只能由拥有者访问。私有数据集同时也能够由其用户设置进行共享。每个注册用户拥有个性化的工作中心,并带有数据管理和数据库工具进行元基因组群落结构等分析。
2.2.3 工作中心
Meta-Mesh的工作中心是一个提供线分析的集成的元基因组计算平台,使用户能够上传数据,进行微生物群落结构分析,样本的比较和搜索等。所有工作中心的功能并不局限于Meta-Mesh数据库,这意味着Meta-Mesh同样能处理用户上传的数据。Meta-Mesh平台的各个工具为元基因组数据分析提供了完整的一站式解决方案。
在Meta-Mesh工作中心,用户可以将分析任务提交到服务器的作业队列,也可以预定任务,服务器将处理数据并在任务结束后把结果返回给用户。服务器后台的作业队列具有自恢复策略,保证用户的数据和分析结果完好无损。工作中心具有以下元基因组相关数据分析工具:
1) 微生物群落结构分析。Meta-Mesh利用Parallel-META[30]分析软件 (工作原理见2.1.2)支持对上传的元基因组数据进行群落样本的生物分类学和进化分析,并具有可配置参数 (图3 C,具体可参考附件1)。Meta-Mesh提供了每个样本的群落结构可视化,网上提供浏览和下载(图3 D)。由于之后的深入分析 (在下面的章节介绍) 依赖于这些结果,Meta-Mesh的微生物群落结构分析被视为一个元基因组样本的“预处理”。
2) 数据上传。Meta-Mesh支持原始序列数据和Parallel-META[30]软件分析产生的群落结构结果的上传。Meta-Mesh接受FASTA或FASTQ格式的原始的元基因组shotgun序列或16S rRNA序列。同时,Meta-Mesh也支持Parallel-META 软件的样本群落分析结果。由于其分析结果是纯文本格式,拥有相对小的尺寸,便于更快上传。
3) 基于生物分类学 (Taxonomy) 的多样本对比。能够从不同生物学层次 (从门 (Phylum)到种 (Species)) 比较两个或更多的样本,并成一个树状分类学视图 (图3E)。图中每个taxon后面的柱状图代表不同样本在此taxon上的相对丰度。该视图可以从多样本比较的结果中下载获得。
4) 基于进化关系 (Phylogeney) 的样本对比和相似性矩阵。Meta-Mesh提供基于进化树的样本量化比较,基于Meta-Storms[28]引擎的样本相似度打分机制 (此算法由本文作者提出,算法内容可参考文献[28],在两个群落的共同进化树中,综合考虑两个群落之间物种的多样性、丰度以及物种间的进化关系,能够自底向上地精确计算出样本间的相似度 (0%–100%之间)。多个样本之间的相似度比较可以生成其相似性矩阵,并生成矩阵的可视化结果 (图3F)。在矩阵中,每个不同颜色方块代表一个相似值,介于红色和绿色之间渐变:红色表示较高的相似值,绿色表示相似值较低。
5) 样本搜索。用户还可以通过Meta-Storms[28]引擎搜索Meta-Mesh 数据库高度相似的样本,并可自定义配置参数 (图3G和H,具体参数可参考附件2,可在网络版中下载) 。这种新颖的功能有助于识别查询样本,尤其是未知的或不完整信息样本。
2.3 基于Web的API和网络服务
为了确保Meta-Mesh平台可广泛使用,我们为外部访问数据库或调用Meta-Mesh的工具提供了一些基于Web的API、包括XML、JSON和HTTP格式。基于这些API,我们开发了一个Web服务模型。通过谷歌地图API (http://meta-mesh.org/map_view) 实现了一个完整的显示微生物群落和样本全球分布的工具。这个扩展的Web服务以及工作中心提供的强大的数据挖掘能力方便了利用样本注释查询相似性的研究。
2.4 软件和数据库访问
Meta-Mesh可以通过网址http://www.metamesh.org进行访问。Meta-Mesh的FTP服务中含有相关数据库数据可下载进行离线使用,其网址为ftp://ftp.meta-mesh.org。Meta-Mesh的自动数据收集器BEGC-SPIDER网址是http://autoupdate.metasee.org。

图3 Meta-Mesh分析平台Fig. 3 Meta-Mesh analysis platform. (A) Project list. (B) Sample list in a project. (C) Structure analysis configurable parameters. (D) Strucutre analysis results. (E) Taxonomical based comparison, bar charts beside taxa represent the abundance of each sample. (F) Quantitate Phylogenetic based sample comparison, each tile in the heat-map represents a similarity value between two samples. (G) Sample searching configurable parameters. (H) Searching results.
3 结果与分析
本文用“人体微生物群落样本的数据库搜索”与“基于群落相似性网络的样本聚类分析”案例研究说明Meta-Mesh系统在元基因组数据分析中的表现。在案例中,本文进行了两种分析:样本的数据库搜索,以及样本聚类分析。该案例所有的数据与分析结果都保存在Meta-Mesh系统中,并可通过登录系统进行查看 (登录信息参考附件3)。
3.1 人体微生物群落样本的数据库搜索
在此研究中,我们使用了来自两个不同性别宿主的3个不同的身体部位:肠道 (来自粪便)、皮肤 (来自手掌) 和口腔 (来自唾液)[36]的60个相关的微生物群落元基因组样本,并在Meta-Mesh数据库系统中进行快速识别。所有查询样本并未收集在Meta-Mesh数据库中。查询样本按照宿主性别和来源身体部位分成6个数据集 (表1),每个样本对整个数据库进行“样本搜索”,找到排名前10位的最佳匹配。
我们评估了每个查询匹配的样本,并基于它们的元数据计算与相应查询样本之间的关系。结果显示,虽然匹配的样本来自不同项目(图4),大多数查询能够确定预期样本的类型或预期的身体部位 (图5,Dataset MO 为100%,Dataset MG为5%,Dataset MS为84 %,Dataset FO为86%,Dataset FG为100%,Dataset FS 为88%)。例如,相比其他查询数据,Dataset MG和Dataset FG对人体肠道 (如项目“FMP_Twins_Mice”[37],详细信息请参阅附表S3)的匹配率更高。此外,手掌的皮肤数据 Dataset MS和Dataset FS查询结果显示,它们和人体其他区域皮肤也有显著的类似,包括手臂 (12%和11%)、足部 (2%和6%)、膝盖 (4%和3%) 和外耳 (0%和5%)。
同时,我们也考察了不匹配的查询结果,发现Dataset MS中10%和Dataset FS中9%匹配结果的是人类口腔微生物群落,以及9%的Dataset FO匹配结构来自手掌皮肤微生物群落。这主要是由于口腔微生物群落结构和手掌上的皮肤群落非常相似,这已经在之前的研究中观察到[28,36]。
在本文中,我们定义了总体的平均正确识别率计算方法R = NC/ N,其中NC表示从数据库中正确匹配的样本数,N表示数据库中所有的匹配样本总数。本实验中,所有的60个群落的查询样本的平均识别正确率R值为92.17% (详细计算过程参考附表S6)。这表明Meta-Mesh数据库和搜索引擎进行快速的搜索结果是非常可靠的。
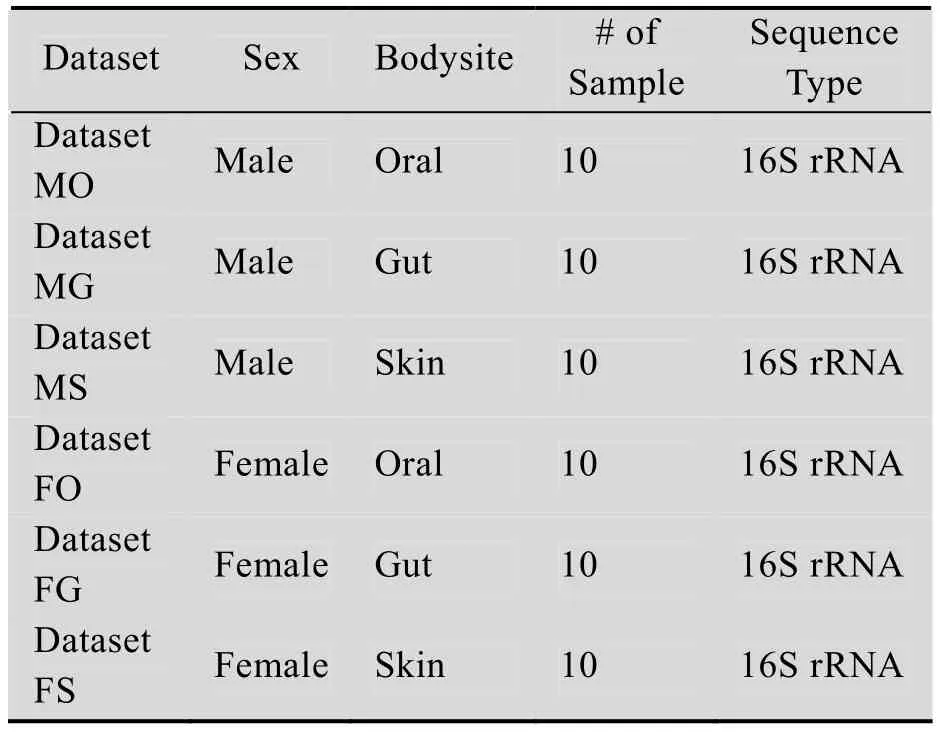
表1 人体微生物群落元基因组样本Table 1 Human-associated habitat metagenomic samples
3.2 基于群落相似性网络的人体微生物群落样本聚类分析

图4 Meta-Mesh数据库中匹配样本项目分布 (项目具体内容可参考附表S3,匹配样本的项目分布具体信息见附表S4)Fig. 4 Projects distribution of matched samples in Meta-Mesh database. The descriptions and details of the projects are included in supplementary Table S3, and details of the matched samples distribution are available in supplementary Table S4.

图5 在Meta-Mesh数据库中的查询搜索结果 (饼状图的每个部分代表数据库的搜索结果,查询样本的身体部位匹配信息见附表S5)Fig. 5 Search results of queries against the Meta-Mesh database. Pie-charts of each body site represent the database search results. Details of the matched results refer to supplementary Table S5.
在本次实验中,我们通过Meta-Mesh计算了本案例中的60个微生物群落样本的相似度矩阵 (图6A),并基于此构建了其相似性网络。在相似性网络中,每个节点代表一个群落样本,而节点之间边的权值即为样本间的相似度。由于在以前的研究中[28]根据P-value值计算,低于85%的相似度即认为是极低的相似性,因此我们在本次实验中将权值低于85%的边进行过滤,并移除孤立点。在构建的相似性网络中,我们采用基于密度的聚类算法MCODE[38]对群落样本进行自然聚类。
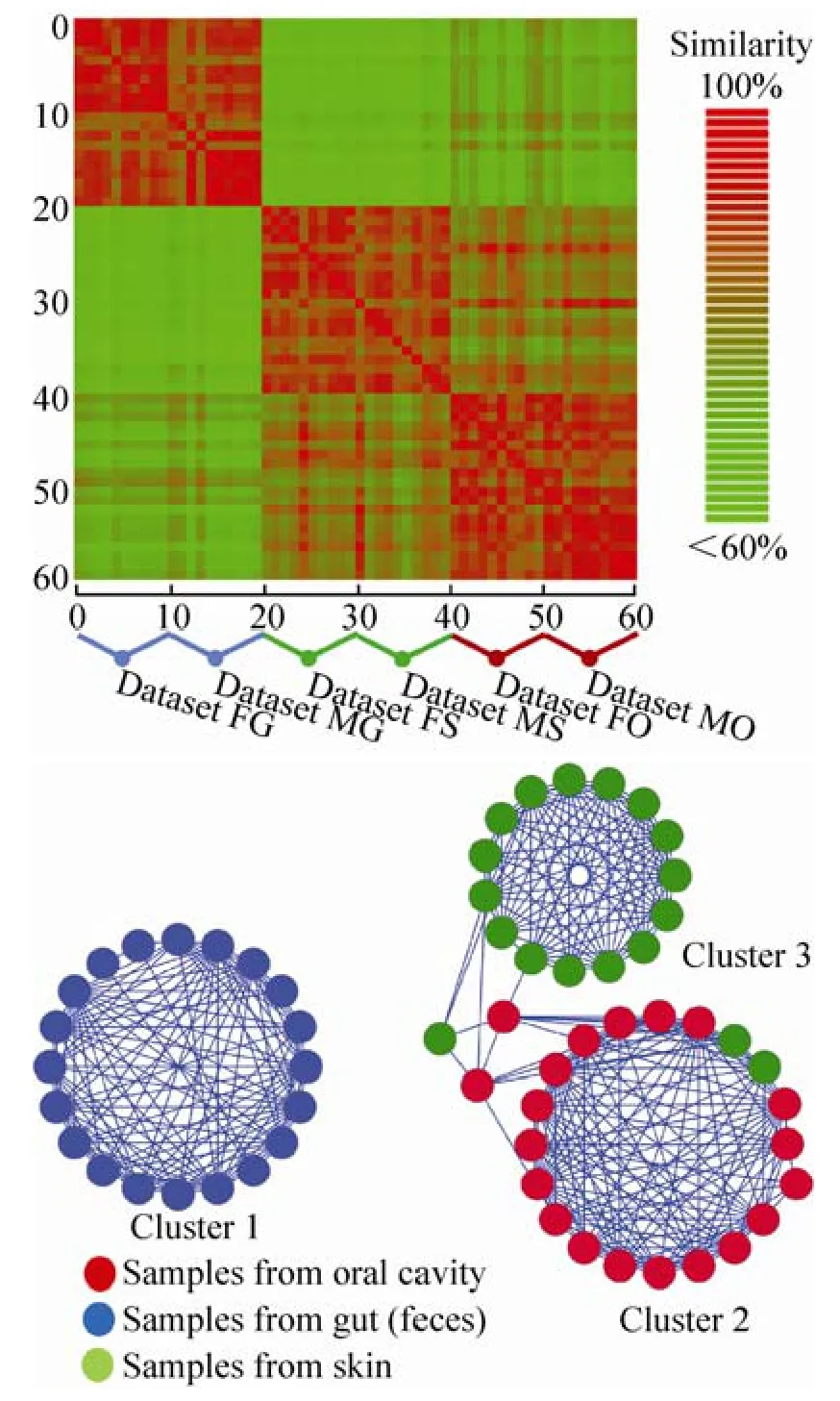
图6 人体微生物群落样本的聚类结果Fig. 6 Clustering results of 60 human-associated habitat samples. (A) Similarity matrix of 60 samples by Meta-Mesh. Blocks represent similarity values between two samples. (B) Clusters by MCODE based on similarity matrix. Nodes represent samples, and edges represent similarity values between samples.
从聚类分析结果 (图6B) 中可观察到,来自相同身体部位的微生物群落被聚类到了一起:这说明了在人体的微生物群落之间,来自不同宿主的相同身体部位的群落结构差异要小于同一宿主不同身体部位的差异。这与之前的多个研究如文献[36]的结果也是相吻合的。
4 讨论和结论
随着微生物群落元基因组研究的扩展和深化,生物信息学分析变得越来越重要。特别是随着新一代高通量测序技术的不断普及,数据量急剧增长;高效率、高精度的生物信息学分析成为了研究微生物群落元基因组的一个核心部分。因此,面向元基因组的重要生物信息学任务之一是:基于超级计算和大规模存储为硬件基础,参考现有的元基因组分析方法和流程,构建高可靠性的元基因组数据分析框架。基于此框架,结合公开和自有的微生物群落高通量测序数据,为微生物群落模式体系 (简单到复杂)归纳测序和分析策略,推动微生物群落数据分析的广泛开展和深入研究。
元基因组样本和序列数据的快速积累,元基因组样本的高效率的比较和元基因组样本库检索已经成为了当前研究中的迫切需求。然而目前缺乏一个好的集成化系统用于组织、比较和数据挖掘。当前的元基因组样本比较主要基于成对的比较 (这使得它们难以进行大规模的分析) ,不支持有效的数据索引。Meta-Mesh克服了这一缺点,是一个集成化的平台,能有效地进行元基因组样本比较分析和数据库检索系统。
通过Meta-Mesh平台对不同的元基因组样本进行比较分型案例研究表明,Meta-Mesh能够准确有效地识别类似的群落结构的样本,开启了研究微生物群落功能多样性的新思路。随着元基因组样本的全基因组测序 (WGS) 的进步,海量的元基因组样本的分析和比较将变得越来越重要,而一个综合比较系统将有巨大的作用。元基因组数据的生物信息学分析目前也已经进入了“大数据”时代。因此,Meta-Mesh定位与此为元基因组学的研究提供关键的方法,从而更广泛进行各种微生物群落的深入数据挖掘,如人类微生物群落项目和地球微生物群系项目等。
Meta-Mesh系统将不断更新,数据库将不断提供高品质的元基因组数据,确保用户可以获取到高质量的实验数据。一些新的数据挖掘技术将进一步提高数据分析的效率,并提供更准确的注释。目前Meta-Mesh数据库是一个基于群落结构的样本分析和相似性检索系统,而基于微生物群落功能分析的平台正在开发中。
随着我们的数据库以及网络服务模块的持续更新、如Parallel-META[30]、 Meta-Storms[28],Meta-Mesh也会同步更新以保持最新的版本。此外,为兼容功能注释数据库,功能结构的分析和比较方法也将被集成到数据分析平台中。
REFERENCES
[1] Proctor GN. Mathematics of microbial plasmid instability and subsequent differential growth of plasmid-free and plasmid-containing cells, relevant to the analysis of experimental colony number data. Plasmid, 1994, 32(2): 101–130.
[2] National Research Council (U.S.). Committee on Metagenomics: Challenges and Functional Applications. and National Academies Press (U.S.), The new science of metagenomics : revealing the secrets of our microbial planet. 2007, Washington, DC: National Academies Press. xii, 158.
[3] Jurkowski A, Reid AH, Labov JB, Metagenomics: a call for bringing a new science into the classroom (while it's still new). CBE Life Sci Educ, 2007, 6(4): 260–265.
[4] Magurran A. Measuring Biological Diversity. Oxford, UK: Blackwell, 2004.
[5] Ley RE, Lozupone CA, Hamady M, et al. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol, 2008, 6(10): 776–788.
[6] Turnbaugh PJ, Ley RE, Mahowald MA, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature, 2006, 444(7122): 1027–1031.
[7] Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature, 2009, 457(7228): 480–484.
[8] Meyer F, Paarmann D, D'Souza M, et al. The metagenomics RAST server-a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics, 2008, 9: 386.
[9] Huson DH, Auch AF, Qi J, et al. MEGAN analysis of metagenomic data. Genome Res, 2007. 17(3): 377–386.
[10] Mitra S, Gilbert JA, Field D, et al. Comparison of multiple metagenomes using phylogenetic networks based on ecological indices. ISME J, 2010, 4(10): 1236–1242.
[11] Mitra S, Klar B, Huson DH. Visual and statistical comparison of metagenomes. Bioinformatics, 2009, 25(15): 1849–1855.
[12] Mou X, Sun S, Edwards RA, et al. Bacterial carbon processing by generalist species in the coastal ocean. Nature, 2008, 451(7179): 708–711.
[13] Kim BH, Chang IS, Gadd GM. Challenges in microbial fuel cell development and operation. Appl Microbiol Biotechnol, 2007, 76(3): 485–494.
[14] Levin DB, Zhu H, Beland M, et al. Potential for hydrogen and methane production from biomass residues in Canada. Bioresour Technol, 2007, 98(3): 654–660.
[15] Etchebehere C, Errazquin I, Barrandeguy E, et al. Evaluation of the denitrifying microbiota of anoxic reactors. FEMS Microbiol Ecol, 2001, 35(3): 259–265.
[16] Harms G, Rabus R, Widdel F. Anaerobic oxidation of the aromatic plant hydrocarbon p-cymene by newly isolated denitrifying bacteria. Arch Microbiol, 1999, 172(5): 303–312.
[17] Gupta R, Beg QK, Lorenz P. Bacterial alkalineproteases: molecular approaches and industrial applications. Appl Microbiol Biotechnol, 2002, 59(1): 15–32.
[18] Backhed F, Ding H, Wang T, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA, 2004, 101(44): 15718–15723.
[19] Backhed F, Manchester JK, Semenkovich CF, et al. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci USA, 2007, 104(3): 979–984.
[20] Kristiansson E, Hugenholtz P, Dalevi D. ShotgunFunctionalizeR: an R-package for functional comparison of metagenomes. Bioinformatics, 2009, 25(20): 2737–2738.
[21] Parks DH, Beiko RG. Identifying biologically relevant differences between metagenomic communities. Bioinformatics, 2010, 26(6): 715–721.
[22] Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol, 2009, 75(23): 7537–7541.
[23] Goll J, Rusch DB, Tanenbaum DM, et al. METAREP: JCVI metagenomics reports--an open source tool for high-performance comparative metagenomics. Bioinformatics, 2010, 26(20): 2631–2632.
[24] Hamady M, Knight R. Microbial community profiling for human microbiome projects: tools, techniques, and challenges. Genome Res, 2009, 19(7): 1141–1152.
[25] Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol, 2005, 71(12): 8228–8235.
[26] Hamady M, Lozupone C, Knight R. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J, 2010, 4(1): 17–27.
[27] Graham CH, Fine PV. Phylogenetic beta diversity: linking ecological and evolutionary processes across space in time. Ecol Lett, 2008, 11(12): 1265–1277.
[28] Su X, Xu J, Ning K. Meta-Storms: efficient search for similar microbial communities based on a novel indexing scheme and similarity score for metagenomic data. Bioinformatics, 2012.
[29] Zhou Q, Su X, Wang A, et al. QC-Chain: fast and holistic quality control method for next-generation sequencing data. PLoS ONE, 2013, 8(4): e60234.
[30] Su X, Xu J, Ning K, Parallel-META: efficient metagenomic data analysis based on high-performance computation. BMC Systems Biology, 2012, 6(Suppl 1): S16.
[31] Mukherjee S, Mitra S. Hidden Markov Models, grammars, and biology: a tutorial. J Bioinform Comput Biol, 2005, 3(2): 491–526.
[32] DeSantis TZ, Hugenholtz P, Larsen N, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB.Appl Environ Microbiol, 2006, 72(7): 5069–5072.
[33] Cole JR, Wang Q, Cardenas E, et al. The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res, 2009, 37(Database issue): D141–145.
[34] Pruesse E, Quast C, Knittel K, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res, 2007, 35(21): 7188–7196.
[35] Griffen AL, Beall CJ, Firestone ND, et al. CORE: a phylogenetically-curated 16S rDNA database of the core oral microbiome. PLoS ONE, 2011, 6(4): e19051.
[36] Caporaso JG, Lauber CL, Costello EK, et al. Moving pictures of the human microbiome. Genome Biol, 2011, 12(5): R50.
[37] McNulty NP, Yatsunenko T, Hsiao A, et al. The impact of a consortium of fermented milk strains on the gut microbiome of gnotobiotic mice and monozygotic twins. Sci Transl Med, 2011, 3(106): 106ra106.
[38] Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics, 2003, 4: 2.
(本文责编 陈宏宇)
Meta-Mesh: metagenomic data analysis system
Xiaoquan Su, Baoxing Song, Xuetao Wang, Xinle Ma, Jian Xu, and Kang Ning
Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, Shandong, China
With the current accumulation of metagenome data, it is possible to build an integrated platform for processing of rigorously selected metagenomic samples (also referred as “metagenomic communities” here) of interests. Any metagenomic samples could then be searched against this database to find the most similar sample(s). However, on one hand, current databases with a large number of metagenomic samples mostly serve as data repositories but not well annotated database, and only offer few functions for analysis. On the other hand, the few available methods to measure the similarity of metagenomic data could only compare a few pre-defined set of metagenome. It has long been intriguing scientists to effectively calculate similarities between microbial communities in a large repository, to examine how similar these samples are and to find the correlation of the meta-information of these samples. In this work we propose a novel system, Meta-Mesh, which includes a metagenomic database and its companion analysis platform that could systematically and efficiently analyze, compare and search similar metagenomic samples. In the database part, we have collected more than 7 000 high quality and well annotated metagenomic samples from the public domain and in-house facilities. The analysis platform supplies a list of online tools which could accept metagenomic samples, build taxonomical annotations, compare sample in multiple angle, and then search for similar samples against its database by a fast indexing strategy and scoring function. We also used case studies of “database search for identification” and “samples clustering based on similarity matrix” using human-associated habitat samples to demonstrate the performance of Meta-Mesh in metagenomic analysis. Therefore, Meta-Mesh would serve as a database and data analysis system to quickly parse and identify similar metagenomic samples from a large pool of well annotated samples.
microbial community, metagenome, database, data mining, online service, similarity network
August 22, 2013; Accepted: November 21, 2013
Kang Ning. Tel: +86-532-80662624; E-mail: ningkang@qibebt.ac.cn
Supported by: Chinese Academy of Science e-Science Program (No. INFO-115-D01-Z006), National High Technology Research and Development Program of China (863 Program) (Nos. 2009AA02Z310, 2012AA02A707, 2014AA021502), National Natural Science Foundation of China (Nos. 61103167, 31271410, 61303161).
中国科学院e-Science项目 (No. INFO-115-D01-Z006),国家高技术研究发展计划 (863计划) (Nos. 2009AA02Z310, 2012AA02A707, 2014AA21502),国家自然科学基金 (Nos. 61103167, 31271410, 61303161) 资助。
